Congenital Disorders of the Lower Airways and Mediastinum
Andrew Bush
Antenatal ultrasound diagnoses many congenital lung malformations, but we now have to decide what to do for a baby affected by an abnormality that may previously have escaped detection. Initial reports described a poor outcome for fetuses with lung masses detected in the second trimester. It is now clear that many lung lesions disappear or regress considerably by term, and the outcome for such fetuses is in general very good. Postnatal regression of lung malformations has also been described. The dilemma in the postnatal period is whether to opt for observation or surgery.
DESCRIPTION OF DISORDERS
The nomenclature of congenital lung disease may be confusing. For example, sequestration and cystic adenomatoid malformation (CCAM), sometimes assumed to be separate identities, but histologic features of both may be found within the same lesion. There is inconsistent use of nomenclature before and after birth. It is necessary to think in new ways about congenital lung disease. The following principles should be followed1:
1. What is actually seen should be described, without embryologic or pathologic speculation, which may later be proved wrong. The same clinical appearance (eg, a multicystic mass) may have one of several different histologic appearances when excised. A simple “catch-all” term, congenital thoracic malformation (CTM), is useful for clinical discussions, prior to any pathologic examination.
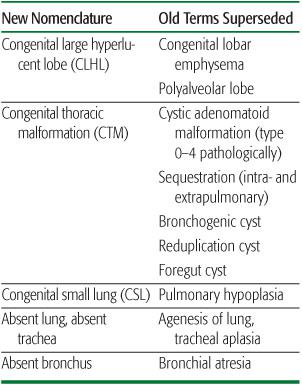
2. The description should be in everyday language, avoiding ambiguity. For example, hypoplastic lung could mean a lung that is small but otherwise normal, or small and structurally abnormal; congenital small lung (CSL) is clearer, and avoids assumptions about structure. Congenital lobar “emphysema” is another source of confusion as in adults it implies lung destruction. What is actually seen is a congenital large hyperlucent lobe (CLHL). Throughout this chapter, established terms will be given in brackets after the proposed new term. A summary comparison of old and new nomenclature is given in Table 507-1, and relevant investigations summarized in Table 507-2.
Table 507-1. Comparison of Present and Past Terms Used to Describe Clinical Appearances of Congenital Lung Malformations
3. The respiratory system should be described systematically. The lung is formed from 6 “trees”: bronchial, systemic and pulmonary arterial, systemic and pulmonary venous, and lymphatic. There are no known abnormalities of bronchial venous drainage, so in practice, only five trees need to be considered.
4. Important associated organs (in particular, the heart, great vessels, chest wall, and abdominal contents) should be considered in a systematic manner. Congenital multisystem diseases may affect the lung.
Pathologic descriptions should also describe what is actually seen (eg, epithelial and mesenchymal components of the mass). Finally, a specific diagnosis may be given, but even after pathologic examination, embryologic speculation is best avoided.
AGE-RELATED PRESENTATIONS
Age-related presentations of congenital lung disease are summarized in eTable 507.1  . Antenatal presentation is usually with either a focal abnormality or abnormal amniotic fluid volume detected by antenatal ultrasound. Absence or reduced fetal breathing movements may be noted, for example in antenatal onset of severe spinal muscular atrophy (SMA). Anything interrupting neural traffic from centrally to the peripheral myocyte that prevents normal respiratory movements will lead to bilateral congenital small lungs (CSLs). Other conditions relevant to the lungs, which may be diagnosed antenatally, include short limbs in skeletal dysplasias associated with bilateral CSL secondary to a small thoracic cavity.
. Antenatal presentation is usually with either a focal abnormality or abnormal amniotic fluid volume detected by antenatal ultrasound. Absence or reduced fetal breathing movements may be noted, for example in antenatal onset of severe spinal muscular atrophy (SMA). Anything interrupting neural traffic from centrally to the peripheral myocyte that prevents normal respiratory movements will lead to bilateral congenital small lungs (CSLs). Other conditions relevant to the lungs, which may be diagnosed antenatally, include short limbs in skeletal dysplasias associated with bilateral CSL secondary to a small thoracic cavity.
Postnatal presentation of a lesion missed antenatally is less common. Primary large airway obstruction presents as stridor, failure to pass an endotracheal tube (ETT), or successful passage of the ETT but inability to establish ventilation immediately after birth. Typically, tracheoesophageal fistula presents as a child choking on feeding, not swallowing saliva, and in whom a nasogastric tube cannot be passed. Respiratory distress in the absence of major airway disease may be due to disorders of the lung parenchyma, including a large cystic or solid congenital thoracic malformation (CTM); unilateral or bilateral CSL; and unilateral or bilateral congenital pleural effusion or lymphatic disorder. Congenital vascular abnormalities may present at this time, including alveolar-capillary dysplasia (ACD), congenital alveolar dysplasia (CAD) spectrum, and pulmonary arteriovenous malformation (PAVM, see below). PAVMs usually present as cyanosis in a well baby (not with heart failure, unless there is an associated systemic, particularly cerebral, arteriovenous malformation). Chest wall disease may present as respiratory distress with difficulty in establishing ventilation. Neuro-muscular disease is usually characterized by inadequate respiratory effort with ease of establishing ventilation, unless there are associated severe unilateral or bilateral CSL.
Table 507-2. Summary of the Role of Investigations in the Management of Congenital Lung Disease*
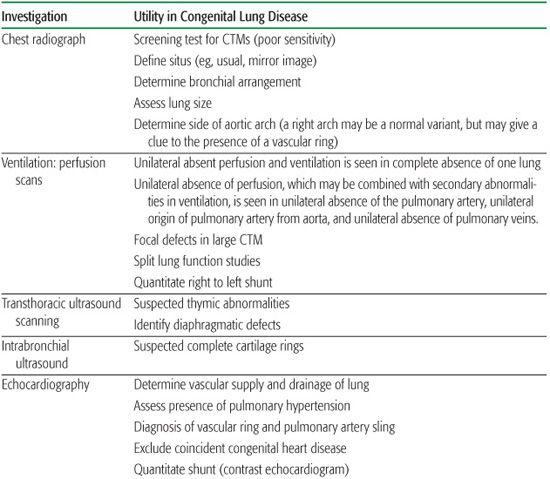
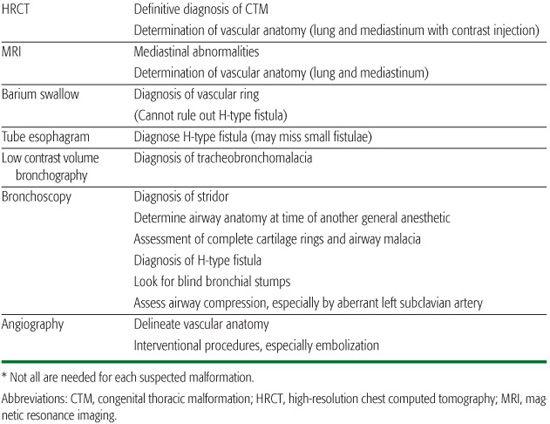
Congenital lung disease may present later in childhood and in adults. Presentation with respiratory distress is rare after infancy, and usually means there is an acute increase in size of the malformation due to bleeding, for example, or rupture causing pneumothorax. CTMs can present as an asymptomatic radiologic abnormality. Unilateral CSL may also be a chance finding. A cystic CTM enters the differential diagnosis2 of recurrent pneumonia in the same location; atelectasis due to large airway compression, dysphagia, lung abscess, focal bronchiectasis, pneumothorax, hemoptysis or hemothorax, or rarely malignant transformation (see below). Complications are unusual in the first 2 years of life. Abnormalities characterized by abnormal systemic arterial supply, unilateral obstruction to pulmonary venous drainage, and PAVM may present with hemoptysis. The latter may also present with progressive cyanosis in a well person, which may lead to polycythemia, or with systemic abscess or embolism, including acute cerebral disease, due to bypass of the pulmonary vascular filter. Large airway narrowing such as tracheomalacia, vascular rings and slings, or complete cartilage rings may present as “steroid-resistant asthma.” Tracheoesophageal fistula may present late, with recurrent bouts of coughing after drinking or hemoptysis.
ANTENATAL DIAGNOSIS AND MANAGEMENT
A fetal lung lesion is suspected either when a thoracic mass is identified or because of mediastinal shift. Investigation may result in fetal therapy being offered or identification of fetuses that should be delivered in a center offering high-level neonatal intensive care and early postnatal surgery. Most abnormalities are detected after approximately 20 weeks of gestation, but some, including diaphragmatic herniae and pleural effusions, may not be detected unless a third trimester scan is performed, or until after birth. Imaging cannot provide a definitive diagnosis for many anomalies. The natural history of many lesions is unknown, and therefore optimal management advice is difficult. Antenatal ultrasound features that were predictive of hy-drops were MTR (ratio of mass size to thorax size), cystic predominance, and diaphragmatic eversion; these fetuses need careful follow-up.
The ultrasonographer should describe the lesion as either macro- or microcystic, not using the term cystic adenomatoid malformation (CCAM)3; cystic lesions may be confused with a diaphragmatic hernia. They usually occur alone, although other associated abnormalities have been described. The prognosis for a fetus with a congenital thoracic malformation (CTM) is generally good; the lesion tends to peak in size at 25 weeks, and then regresses.4 Polyhydramnios and mediastinal shift were considered indicators or poor prognosis, but more recent data suggest that these are less reliable indicators of outcome than once thought. Accurate prediction of outcome for prenatally diagnosed lesions can be difficult following a single scan, and serial scans should be performed. Early consultation with neonatal and pediatric surgical staff is helpful for parents. Where there are single or multiple large cysts with associated hydrops or polyhydramnios improvement has been reported with in utero decompression by thoracocentesis, the insertion of a shunt or even surgical resection However, because many lesions spontaneously regress, fetal intervention should be seen as a last resort. A so-called fetal sequestrated lobe is most often identified as an echogenic mass of uncertain origin in the chest or subdiaphragmatic area. Prenatally it is not possible to make a definitive diagnosis even if an independent blood supply is demonstrated, since a CCAM may also have an aortic blood supply. This underscores the value of the generic term CTM. Where any lesion has persisted or increased in size and mediastinal shift persists in the third trimester, delivery in a centre with neonatal intensive care and surgical facilities is advisable. All infants in whom any suggestion of a lung malformation has been made antenatally will require a chest radiograph (CXR) prior to discharge. In one study of 19 infants, CXR was only 61% sensitive for malformations, whereas CT, the gold standard, was 100% sensitive and specific.5
POSTNATAL FEATURES
 AIRWAY AND LUNG PARENCHYMA
AIRWAY AND LUNG PARENCHYMA
Congenital Abnormalities of the Trachea
Tracheomalacia leads to tracheal collapse due to localized or generalized weakness of the tracheal wall, and respiratory obstruction (see Chapters 371 and 510).  Tracheal atresia is rare; isolated atresia may be amenable to surgical reconstruction, but most are fatal. Minor tracheal hypoplasia and other airway abnormalities, including complete cartilage rings, may be seen in Down syndrome.7
Tracheal atresia is rare; isolated atresia may be amenable to surgical reconstruction, but most are fatal. Minor tracheal hypoplasia and other airway abnormalities, including complete cartilage rings, may be seen in Down syndrome.7
Stenosis usually occurs as because of a web in the subglottic area or just above the carina, although diffuse involvement has also been recorded. An abnormally long and funnel-shaped trachea may also occur with complete cartilage rings, which may be associated with pulmonary artery sling.8,9 This diagnosis may be confirmed by bronchoscopy, endobronchial ultrasound, MRI, or low contrast volume bronchography. If the tracheal lumen is small this may be incompatible with survival. In milder cases, there may be respiratory distress in the delivery room unrelieved by intubation or even tracheostomy. Milder cases may require no treatment, and often tracheal growth keeps pace with the child. Surgery may become necessary as the child grows, if exercise tolerance is severely limited. Options include resection or reconstruction of the stenotic segment, or even tracheal transplant for really severe and complex cases. The multiplicity of surgical techniques reflects the relatively poor outcome in the severely symptomatic.
Congenital Abnormalities of the Bronchial Tree
Congenital bronchial stricture occurs predominantly in a main stem or middle-lobe bronchus and can lead to acute and/or chronic pulmonary infection. Inflammatory scarring of the congenitally stenosed bronchus leads to distal suppuration, atelectasis, and bronchiectasis. Atresia is usually asymptomatic and detected incidentally on x-ray. The airway may be blocked by a simple membrane, or there may be a discontinuity. Atresia often results in cystic degeneration of the lobe distal to the obstruction before birth, as fetal lung liquid continues to be secreted and cannot drain. Absent bronchus and bronchogenic cyst may be more common than was once thought (see below).
Abnormal Bronchial Origin and Bronchial Branching
Bronchi can arise from the gastrointestinal tract. The right upper lobe bronchus can arise from the trachea (“pig bronchus”). This is usually of no clinical significance, but may be a cause of recurrent right upper lobe collapse in an intubated patient if the endotracheal tube (ETT) is low. The right lower lobe bronchus may also arise from the left bronchial tree, a so-called “bridging bronchus.” Whole lung segments may also cross over.
Disorders of Bronchial Laterality
The two most useful determinants of right lung morphology are the presence of three not, two lobes, and a very short main bronchus prior to the takeoff of the upper lobe. Mirror-image arrangement must be distinguished from congenitally small (hypoplastic) right lung with right sided heart (dextroposition). Mirror-image arrangement and other forms of heterotaxy10 may be a feature of primary ciliary dyskinesia, whereas a congenital small lung needs to have its vascular supply delineated (below). The word isomerism is so entrenched that it is probably not feasible to replace it with the term bilateral right lung), which might be simpler to understand. Nearly 80% of children with right isomerism, i.e., bilateral right lung, have asplenia and are thus at risk of overwhelming pneumococcal sepsis. Ivemark syndrome consists of right isomerism, asplenia, a midline liver, malrotation of the gut, and a variety of cardiac abnormalities.11 Left isomerism, i.e. bilateral left lung, is associated with polysplenia in around 80% of patients. These patients may also have a midline liver, malrotation of the gut, partially anomalous pulmonary venous drainage, and cardiac septal defects.12
Disorders of the Bronchial Walls
All or part of the bronchial wall caliber may be too large or too small. These may present with recurrent infections, steroid unresponsive wheeze, or stridor. Congenital tracheobronchomegaly (Mounier-Kuhn syndrome) is characterized by tracheomalacia and greatly dilated major airways. True congenital bronchiectasis is much rarer than previously thought.
The bronchial lumen may be narrowed by complete cartilage rings. There may be an associated pulmonary artery sling. A short segment may require no treatment. If ventilation is critically compromised, surgical excision or a Z-plasty may be indicated. Congenital bronchomalacia may be isolated, often with a good prognosis, or associated with other congenital abnormalities.
 PULMONARY AGENESIS, APLASIA (ABSENT LUNG), AND HYPOPLASIA (SMALL LUNG)
PULMONARY AGENESIS, APLASIA (ABSENT LUNG), AND HYPOPLASIA (SMALL LUNG)
Bilateral absent lungs is a rare malformation that may occur in anencephaly. Unilateral absent lung is slightly more common. Lobar agenesis and aplasia are rarer than complete absence of one lung and usually affect the right upper and middle lobes together. Pulmonary hypoplasia (CSL) consists of incompletely developed lung parenchyma connected to bronchi that may also be underdeveloped dependent upon when the presumed casual insult took effect in embryogenesis. There are a large number of causes of CSLs.  Children with complex malformations, for example unilateral CSL with abnormal vasculature, may be asymptomatic for long periods despite a formidable list of abnormalities. Any aortopulmonary collaterals should be occluded. Long survival, even with an absent lung, is quite possible.
Children with complex malformations, for example unilateral CSL with abnormal vasculature, may be asymptomatic for long periods despite a formidable list of abnormalities. Any aortopulmonary collaterals should be occluded. Long survival, even with an absent lung, is quite possible.
 CONGENITAL CYSTIC LESIONS
CONGENITAL CYSTIC LESIONS
Numerous pathologic conditions present with cystic changes in the lung, with acquired lesions outnumbering those that are developmental in origin. It has been recognized pathologically that bronchial atresia is a common accompaniment of congenital thoracic malformations (CTMs). Presentation of these lesions has been discussed above. This section discusses the pathology. It is usually only after surgical excision that sensible pathologic diagnoses can be made.
Foregut (Bronchogenic) Cysts (Clinically, Cystic CTM)
Foregut cysts are epithelial-lined sacs in the thorax. Bronchogenic cysts, distinguished by the presence of cartilage in the wall are the most common cysts reported in infancy, although many do not present until adulthood. About 50% are situated in the mediastinum close to the carina, and less frequently adjacent to the oesophagus and alongside the tracheobronchial tree. More rarely, they are found within the lung parenchyma and exceptionally they are extrathoracic. Foregut cysts are usually single, unilocular, and more common on the right. They may have a systemic blood supply. Fistulous connections between cysts and the bronchial tree have been reported. Malignant transformation is exceptionally rare but is reported in gastroenteric cysts.
Cystic Adenomatoid Malformation
Cystic adenomatoid malformations (CCAMs) encompass a spectrum of variably sized cysts with differing histology. The reported incidence is between 1:25000 and 1:35000, although antenatal ultrasound is causing us to increase estimations of prevalence. Type 0 CCAMs, also termed acinar dysplasia, are rare, incompatible with life, and typically associated with other abnormalities. Type 1 is the most common type of CCAM, and has the best prognosis; cysts are over 2 cm in diameter by definition. The cystic spaces are lined by pseudostratified ciliated columnar epithelium and mucus cell hyperplasia is seen in 35% to 50% of cases. Type 2 CCAMs are the second most frequent type. They generally cause respiratory distress in the first month of life and may be associated with renal agenesis, cardiovascular defects, diaphragmatic hernia, and syryngomyelia. Macroscopically, the lesions are multiple small cysts. Microscopically the cystic airspaces relate to a relative overgrowth of dilated bronchiolar structures that are separated by alveolar tissue, which appears comparatively underdeveloped. Type 3 CCAMs are uncommon and occur almost exclusively in males. They typically involve and expand a whole lobe, the others being compressed. Macroscopically, lesions appear solid and not cystic. Microscopically, there is an excess of bronchiolar structures separated by air spaces that resemble late fetal lung. Type 4 CCAMs are also very rare and comprise peripheral thin-walled cysts that are often multiloculated. The cystic spaces are typically lined by alveolar type I or type II cells with the intervening stroma being thin and comprising loose mesenchymal tissue. There is overlap with type 1 pleuropulmonary blastomas.
Treatment of Congenital Cystic Lesions
If a cystic lesion is causing significant symptoms, usually during the newborn period, despite medical therapy, then the baby needs surgery. For all but the smallest, sickest infants, lobectomy (usually thoracoscopic) is a safe and well-tolerated procedure, with few if any significant sequelae. Pneumonectomy carries a significant mortality in infancy, and long-term morbidity, in particular scoliosis, which may worsen during the pubertal growth spurt.
If a previously asymptomatic cystic lesion has become infected, the lesion should be excised. Other unequivocal indications for treatment, usually surgery or coil embolization of a feeding vessel, are respiratory distress and failure to thrive, usually in the newborn period; high output heart failure due to aortopulmonary collateral flow; pneumothorax nonresponsive to conventional therapy; infection of the malformation, or chronic distal infection due to airway compression, and uncontrolled hemorrhage from the malformation (hemothorax or hemoptysis).2 The management of the small, asymptomatic congenital thoracic malformation (CTM) is controversial. Some operate on all but the tiniest malformations, and others only operate on symptomatic babies. Proponents of neither view can adduce convincing evidence for their position. If the lesion is cystic, then it is likely (but unproven) that infection is likely sooner or later, and prophylactic excision would be recommended by many.13 It is said that all lesions should be excised to prevent malignant transformation, but there is little evidence in favor of this view (see below). Whether an asymptomatic malformation should be resected to prevent the development of complications, or to facilitate normal lung growth, is not clear. If the risk of these lesions becoming infected is unknown, we know even less about the risk of malignant transformation. Primary pulmonary malignancy in childhood is very rare, but there are reports of coexistence of CTM and a variety of primary pulmonary malignancies. Removal of a CTM cannot prevent the development of malignant disease elsewhere in the lung,14 implying that the malformation is merely a marker of generalized increased malignant potential. There is insufficient evidence on which to base recommendations as to how to prevent malignancy.
If surgery is contemplated, it is essential to delineate the anatomy of the congenital thoracic malformation (CTM), including the blood supply. Contrast high-resolution chest computed tomography (HRCT) gives the best images of parenchymal abnormalities, and is currently probably the investigation of choice in suspected CTM.
 PULMONARY SEQUESTRATION
PULMONARY SEQUESTRATION
The classic definition is tissue that is isolated from normal functioning lung and is nourished by systemic arteries, although the separation may be purely vascular. The intrapulmonary variant is contained within otherwise normal lung parenchyma. The less common extralobar sequestration is accessory to the lung. Sequestrations may also connect to the oesophagus or stomach, as well as contain pancreatic tissue and also may show histologic features of cystic adenomatoid malformation (CCAM). The etiology is not understood. Intralobar sequestrations are usually found in the posterior basal segment of the left lower lobe and extralobar sequestrations beneath the left lower lobe. About 15% of extralobar sequestrations are abdominal. The intralobar sequestration is encircled by visceral pleura and has no pleural separation from the rest of the lobe. The remainder of the affected lobe and lung is normal, unless secondary changes such as infection have supervened. More than half the cases of intralobar sequestration are diagnosed after adolescence, and symptoms in neonates and infants are uncommon. Extralobar sequestration is generally detected in infancy because of associated malformations and affects males four times more frequently than females. Although much rarer, intralobar sequestrations may also be associated with other malformations. Treatment of sequestration is surgical excision. The vascular supply should be carefully delineated by preoperative investigations, and embolization of aortopulmonary collaterals may be considered.
 CONGENITAL LARGE HYPERLUCENT LOBE (CONGENITAL LOBAR EMPHYSEMA)
CONGENITAL LARGE HYPERLUCENT LOBE (CONGENITAL LOBAR EMPHYSEMA)
Congenital large hyperlucent lobe (CLHL) is a rare condition that affects the left upper (42%), right middle (35%), right upper (21%), and lower lobes (2%). The affected lobe over-distends and displaces adjacent lobes and subsequently the mediastinum, and may herniate into the contralateral hemithorax. The condition may be diagnosed antenatally; present as respiratory distress, often with consequent failure to thrive in infancy; or be a chance finding on a chest radiograph (CXR) taken later in life. Clinical features of infantile lobar emphysema are suggestive of a tension pneumothorax. Usually, a CXR will demonstrate a hyperlucent lobe with features of compression and collapse of the adjacent lung, ipsilateral depression of the diaphragm, and mediastinal shift. Bronchoscopy may reveal causes of intrinsic obstruction and permit the removal of a foreign body or inspissated secretions, or even provide temporary relief of symptoms to facilitate surgery.15 The differential diagnoses is any intrinsic or extrinsic airway cause of failure of airspace emptying and any cause of loss of lung volume on the contralateral side including absent lung and lobar or lung collapse due to bronchial obstruction. Children who are thriving and asymptomatic require no treatment, otherwise treatment is lobectomy.
 ABNORMAL CONNECTIONS BETWEEN THE BRONCHIAL TREE AND OTHER STRUCTURES
ABNORMAL CONNECTIONS BETWEEN THE BRONCHIAL TREE AND OTHER STRUCTURES
Tracheoesophageal Fistula (TOF) and Esophageal Atresia
The genetics, associated disorders, diagnosis, and treatment of tracheoesphageal fistula is discussed in Chapter 392.
Other Abnormal Connections
Rare direct communications between the bronchial tree and congenital thoracic malformation (CTM), the biliary tract, and the stomach have been described.
 CONGENITAL ABNORMALITIES OF THE PULMONARY ARTERIAL TREE
CONGENITAL ABNORMALITIES OF THE PULMONARY ARTERIAL TREE
Systemic arterial abnormalities can be separated into those of the bronchial circulation; other pathologic collaterals; and airway compression by abnormal great vessels, for example a double aortic arch. The pulmonary capillary bed may be bypassed by direct arteriovenous communication, or absent, resulting in minimal pulmonary arteriovenous connections.
Disorders of Pulmonary Artery Arrangement
In general, pulmonary arterial and venous arrangement mirrors bronchial arrangement. Exceptions include congenital origin of the left pulmonary artery from the right (pulmonary artery sling), where the left pulmonary artery traverses the mediastinum compressing the trachea. There may also be a crossover arterial segment, with the right upper lobe supplied from the left pulmonary artery, or complete cartilage rings. Isolated crossover pulmonary artery branches in the absence of bronchial crossover are occasionally seen (see Chapter 484).
Absent or Small Pulmonary Artery
Absent or small pulmonary artery may be isolated or associated with other cardiovascular anomalies. Symptoms may not arise until adult life, and include pulmonary infection or hemorrhage. Congenitally small unilateral pulmonary artery is usually associated with an ipsi-lateral congenital small lung (CSL). Pulmonary arterial stenosis may affect lobar and segmental vessels as well as the main pulmonary arteries. Isolated unilateral absence of a pulmonary artery may be asymptomatic, or there may be infection or bleeding from bronchopulmonary anastomoses. Anomalous systemic arteries supplying the lung may be associated with any congenital thoracic malformation (CTM), or even be an isolated finding.17 They are also found if the pulmonary artery is absent and may also be part of complex arteriovenous malformations. One or both pulmonary arteries may take origin from the aorta.18 Bilateral origin from the aorta is part of the spectrum of common arterial trunk, and usually will present to the pediatric or fetal cardiologist. Unilateral origin of a pulmonary artery from the aorta may be an isolated abnormality, sometimes presenting with persistent tachypnea.
 RELEVANT CONGENITAL ABNORMALITIES OF THE SYSTEMIC ARTERIAL TREE
RELEVANT CONGENITAL ABNORMALITIES OF THE SYSTEMIC ARTERIAL TREE
Vascular ring causes compression of the tracheo-esophageal complex. Presentation is usually with stridor, or ‘steroid resistant asthma’, but occasionally dysphagia dominates. (See Chapter 484.)
CONGENITAL ABNORMALITIES OF THE PULMONARY VENOUS TREE
Abnormal Pulmonary Venous Drainage
These anomalies are discussed in Chapter 484.
Congenital Absence of the Pulmonary Veins
Absence or narrowing of the pulmonary veins results in pulmonary venous obstruction and pulmonary hypertension. Atresia may be unilateral or bilateral. Prognosis is poor.
 ABNORMALITIES OF THE CONNECTIONS BETWEEN THE PULMONARY ARTERIAL AND VENOUS TREES
ABNORMALITIES OF THE CONNECTIONS BETWEEN THE PULMONARY ARTERIAL AND VENOUS TREES
Disorders of Alveolar Capillary Development
Part of the alveolar capillary dysplasia-congenital alveolar dysplasia (ACD-CAD) spectrum encompasses misalignment of lung vessels, representing a failure of capillaries to grow in appropriate number and location within the alveolar tissue of the lung, often associated with abnormally sited pulmonary veins.
Pulmonary Arteriovenous Malformation
Pulmonary arteriovenous malformations (PAVMs) are abnormal direct connections between the pulmonary arterial and venous trees. These occur most commonly in the lower lobes; about 75% of patients have unilateral lesions and about 36% of patients have multiple lesions, of which about half are bilateral. Many are associated with hereditary hemorrhagic telangiectasia (HHT). Mutations in the endoglin and ALK-1 genes, respectively, cause HHT1 and HHT2.19,20 The fistula is fed by at least one afferent artery, usually pulmonary, and may be drained by several veins, almost always pulmonary. In 80% of patients there is a single feeding pulmonary artery and a single draining pulmonary vein. Abnormal direct connections between the pulmonary arterial and venous trees (PAVMs) may also be diffuse and microscopic. Presentation is with dyspnea, cyanosis, hemoptysis, or neurologic complications, such as cerebral abscess.21 There may be clubbing, cyanosis, and a chest wall bruit. The circulation is hyper-dynamic only if there is a substantial systemic arterial component. The chest radiograph may be normal, but more usually well defined single or multiple opacities are seen, with vessels connecting them to the hilar, which are strongly suggestive of the diagnosis. High-resolution chest computed tomography (HRCT) scanning will show all but diffuse microscopic malformations.22 Diagnosis is suspected on imaging and confirmed by documenting an abnormal intra-pulmonary right-to-left shunt by contrast echocardiography. Treatment is by embolization, which reduces the risk of complications.21 Systemic complications are mainly related to the bypassing of the filtration function of the pulmonary circulation, and hyperviscosity secondary to systemic arterial hypoxaemia. Pulmonary hypertension does not develop, because there is no alveolar hypoxaemia. The patient may develop systemic abscess or thrombosis. Patients with diffuse PAVM are a high-risk group.23 Local complications including hemothorax and massive hemoptysis have been described.24,25
 CONGENITAL ABNORMALITIES OF THE LYMPHATIC TREE
CONGENITAL ABNORMALITIES OF THE LYMPHATIC TREE
Congenital pulmonary lymphangiectasia may be primary to obstruction of pulmonary lymphatic or venous drainage or secondary, either limited to the lung or generalized. It causes severe respiratory distress and is often fatal in the neonatal period, but milder cases with prolonged survival have been described.
 CONGENITAL CARDIAC DISORDERS
CONGENITAL CARDIAC DISORDERS
Cardiac malformations may be coincidental to, or a fundamental part of, a pulmonary malformation (see Chapter 484). Echocardiography should be performed routinely in most suspected congenital lung abnormalities. Vascular compression of the airways in the setting of congenital heart disease is not uncommon.
 MULTISYSTEM CONGENITAL DISORDERS THAT AFFECT THE LUNG
MULTISYSTEM CONGENITAL DISORDERS THAT AFFECT THE LUNG
A few congenital lung abnormalities are part of a generalized disorder, such as tuberous sclerosis, and metabolic diseases, such as Niemann-Pick, and lysinuric acid deficiency.
 LOBECTOMY
LOBECTOMY
Lobectomy is well tolerated, and it is extent, not age at resection that is important. It is unlikely that a large malformation would interfere with the growth of surrounding normal lung. Follow-up of children with a residual congenital thoracic malformation (CTM) is not evidence based.
REFERENCES
See references on DVD.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


