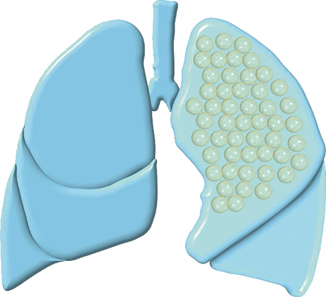
Fig. 51.1
Diagramatic representation of type I CCAM
Multiple large cysts (> 2 cm in diameter).
A single large cyst surrounded by numerous smaller cysts.
Type I is the most common type of CCAM and is associated with an excellent prognosis.
The cysts are frequently lined by pseudostratified columnar epithelial cells, which occasionally produce mucin.
2.
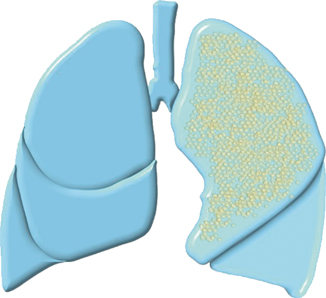
Type II CCAM (25 %; Fig. 51.2):
Fig. 51.2
Diagramatic representation of type II CCAM
This is characterized by multiple small cysts, usually <1 cm in diameter.
Accounts for > 25 % of cases of CCAM.
They are characterized by small, relatively uniform cysts resembling bronchioles.
As many as 60 % of type II lesions are associated with other congenital anomalies that may affect prognosis, specifically renal agenesis.
The cysts generally measure 0.5–2 cm in diameter.
These cysts are lined by cuboid-to-columnar epithelium and have a thin fibromuscular wall.
3.
Type III CCAM (10 %; Fig. 51.3):
Fig. 51.3
Diagramatic representation of type III CCAM
These are large and grossly a solid mass without obvious cyst formation.
They account for < 5–10 % of all cases of CCAM.
Microscopically, they consist of multiple microcysts, measuring < 0.5 cm in diameter.
In 1993, Adzick reported his classification of CCAM into two types.
1.
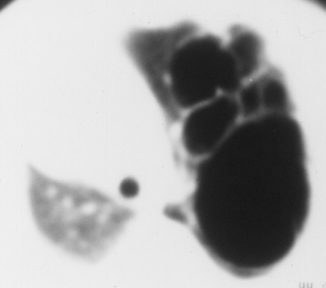
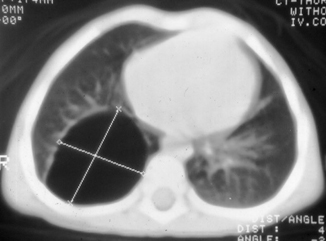
Fig. 51.4
CT scan of the chest showing type I CCAM. Note the large cyst and multiple smaller cysts
Fig. 51.5
CT scan of the chest showing a large cyst (type I CCAM)
Cysts measuring < 5 mm in diameter
Usually associated with fetal hydrops
Have a poor prognosis
2.
Macrocystic CCAM :
Cysts measuring > 5 mm in diameter
Usually not associated with hydrops
Have a good prognosis
CCAM receives its blood supply from the pulmonary circulation and is not sequestered from the tracheobronchial tree.
Microscopically, the lesions are not true cysts, but communicate with the surrounding lung parenchyma.
However, type II and III lesions can occasionally coexist with extralobar sequestration, and in such cases, they may receive systemic arterial blood supply.
CCAM may also occur in association with a polyalveolar lobe.
A polyalveolar lobe is a form of congenital emphysema with increased number of alveoli with normal bronchi and pulmonary vasculature.
CCAM usually occurs early in fetal life, whereas polyalveolar lobe occurs late.
The pathophysiologic effects of CCAM may be divided into prenatal and postnatal effects.
Large lesions may be associated with the development of hydrops fetalis in as many as 40 % of cases and is a poor prognostic sign.
Hydrops is thought to arise from compression of the inferior vena cava, which compromises venous return and leads to a decrease in cardiac output and the development of effusions. Fetal demise may result; premature delivery is attempted in order to salvage the fetus.
The other main prenatal event is compromised pulmonary growth. This results in pulmonary hypoplasia which may lead to postnatal respiratory distress.
Presentation
The usual postnatal presentation of CCAM is a respiratory distress in the newborn period.
This may be due to:
Pulmonary hypoplasia
Mediastinal shift
Spontaneous pneumothorax
Air trapping within the cyst leading to compression of functional pulmonary tissue
Pleural effusion secondary to hydrops
It may range in severity from grunting, tachypnea, and a mild oxygen requirement to fulminant respiratory failure requiring aggressive ventilator support or extracorporeal membrane oxygenation (ECMO).
CCAM may also remain undiagnosed until it is discovered as an incidental finding later in life.
Recurrent chest infections due to bronchial compression, air trapping, and inability to clear secretions may be the presenting feature in those presenting later in life.
A risk of malignant transformation in later years is also noted.
Hemoptysis has occasionally been described as a manifestation of CCAM in older children.
Cough, fever, and failure to thrive have all been reported in association with the presentation of CCAM.
Prenatal regression and complete resolution of CCAM have also been described.
Differential Diagnosis
The differential diagnosis includes:
Congenital diaphragmatic hernia. This is especially so for CCAM affecting the left lower lobe of the lung.
Congenital pneumonia.
Hemothorax.
Pleural effusion.
Pneumatocele.
Pneumothorax.
Pulmonary sequestration.
Pneumatoceles that form subsequent to bacterial pneumonia (e.g., streptococcal, staphylococcal) can be mistaken for CCAM, particularly in the older child.
Congenital lobar emphysema refers to overexpansion of one lobe of the lung, typically an upper lobe or right middle lobe that leads to mass effect and respiratory distress. Although this entity could potentially be confused with CCAM, typical features of overexpanded but normal parenchyma can be observed and confirmed with computed tomography (CT) scan if necessary.
Pulmonary interstitial emphysema may resemble CCAM when it is complicated by large air collections. However, these are also typically associated with linear collections and preceded by high-pressure ventilation and barotrauma. The air collections are located in the interstitial lymphatics.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


