Congenital Central Hypoventilation Syndrome
Epidemiology and History
In 1970, Dr. Robert Mellins described the first congenital case of primary central alveolar hypoventilation, and reviewed 30 postnatally acquired central hypoventilation reported cases in his seminal publication entitled, Failure of Automatic Control of Ventilation.1 Until then, all reported cases of central hypoventilation were considered as acquired, and were attributed to abnormalities of the central nervous system (e.g., encephalitis or focal medullary abnormality). This now classic case report described a newborn boy of normal birth weight, who cried at birth and demonstrated improved ventilation with crying, but was persistently cyanotic while in the nursery, without identifiable etiology after thorough diagnostic work-up. He improved with negative-pressure ventilation therapy while initially hospitalized, and was subsequently surgically implanted with phrenic nerve electrodes stimulated via radiofrequency transmitter (phrenic nerve diaphragm pacing). Since the pacing was unsuccessful, he succumbed to heart failure and died at 14 months of age. Despite the therapeutic failure,1 diaphragmatic pacing in congenital central hypoventilation syndrome (CCHS) has become more refined, takes into consideration multiple factors, and is an ideal method of artificial ventilation in the appropriate CCHS candidate.2 However, the authors provide a compelling description of the physiologic compromise the child suffered during various states, the ventilatory response to exogenous hypercarbic challenge, and also describe some of the sequelae of chronic inadequate ventilatory support.
While many case reports followed, it was not until 1992 that Dr. Weese-Mayer and colleagues described the first relatively large cohort of 32 CCHS cases with description of associated findings (Hirschsprung disease, ophthalmologic abnormalities, growth deficiency) and outcomes.3 In this cohort, the authors found that about one third died, a third required awake and asleep ventilatory support, and the other third required sleep-only ventilatory support. In 1999, the first American Thoracic Society (ATS) statement on CCHS estimated the existence of at least 160–180 cases worldwide.4 Further, this publication increased awareness of this rare condition, introduced the concept of autonomic nervous system (ANS) dysregulation (ANSD), and suggested the existence of a genetic basis for CCHS.4 Subsequently, candidate gene studies focused on the ANS pathways.
A major breakthrough occurred in 2003 with the discovery that PHOX2B gene mutations were present among CCHS patients in French,5 American,6 and Japanese cohorts.7 The PHOX2B gene is located on chromosome 4, is a transcription factor that plays a key role in embryologic development of the autonomic nervous system,8,9 and the genetic abnormality is inherited in an autosomal dominant pattern.6,10 Race and gender are not expected to influence inheritance or severity of illness. The development of clinically available diagnostic testing allowed for earlier and more definitive diagnosis of CCHS with a consequent increase in the number and frequency of identified cases.
In 2010, the second ATS statement on CCHS was published and estimated at least 1000 living children with CCHS worldwide11 and suggested this number to represent an underestimate since those patients with milder CCHS phenotypes are likely to be missed. The 2010 ATS statement also reiterated the clinical criteria for diagnosis of CCHS and introduced the requirement for identification of PHOX2B gene mutations for the definitive diagnosis of CCHS. Over the past decade, mortality and morbidity of CCHS appear to have improved with introduction of genetic testing because of the ability for early, definitive diagnosis that allows for early introduction of chronic artificial ventilatory support, facilitates parental acceptance of diagnosis and provides empowerment in the context of prenatal testing, and allows for anticipatory guidance and insight into prognosis based on specific type of PHOX2B mutation. However, specific studies are lacking to conclusively demonstrate the improved outcomes associated with earlier detection and recognition.
Presentation and Diagnosis
The majority of individuals with CCHS presents in the newborn period with signs of alveolar hypoventilation resulting in hypoxemia and hypercarbia, which are most apparent during sleep, and are usually not accompanied by any associated increases in respiratory rate or arousal.4 CCHS should be considered if an intubated individual requires minimal pressure (suggesting normal lung compliance) and is unable to tolerate decreases in ventilator rate settings and/or experiences unexpected failed extubations associated with absent cardiorespiratory and behavioral responses to ensuing physiologic compromise. PHOX2B genetic testing should be immediately performed while evaluation of other potential etiologies for the alveolar hypoventilation are being pursued – exclusion of pulmonary, cardiac, or neuromuscular disease and brainstem lesions. If feeding intolerance or constipation is present, then evaluation for Hirschsprung’s disease should also be considered. Associated pathology and the spectrum of ANSD can include decreased heart rate variability,12 abrupt sinus pauses,13 decreased pupillary responses to light,14 decreased basal body temperature, esophageal dysmotility, constipation, and altered diaphoresis.4,11,15 These subtle symptoms may be difficult to identify unless specifically sought.
Patients presenting outside of the newborn period are called ‘late-onset’ CCHS (LO-CCHS). Although the delayed diagnosis may be attributable to subclinical rather than absence of symptoms, alveolar hypoventilation often becomes readily apparent in the context of an instigating event, such as exposure to sedation/substances that decrease the level of consciousness or following an acute pulmonary process such as pneumonia that requires an increase in respiratory drive. With careful, comprehensive review of medical history in these individuals, subtle signs and symptoms of disordered respiratory control from infancy may be uncovered.
Paired-Like Homeobox 2B (PHOX2B) and Genetic Testing
The PHOX2B gene consists of three exons; the third exon has two polyalanine repeat regions of which the larger polyalanine repeat region normally has 20 alanines on each allele. Therefore, the PHOX2B genotype in a normal individual would be indicated as ‘20/20.’ Ninety percent of CCHS-associated PHOX2B mutations are due to heterozygous polyalanine repeat expansion mutations (PARM) with expansions from 24 to 33 alanine repeats on the affected allele (resulting genotypes 20/24 – 20/33).11 Of these, the most common PHOX2B PARMs are genotypes 20/25, 20/26, and 20/27.16 PHOX2B mutations that do not consist of polyalanine expansions are referred to as non-polyalanine expansion repeat mutations (NPARM), and may include frameshift, nonsense, and missense mutations. More recently, whole exon deletions have been identified as causing CCHS in <1% of cases, though these cases may just have components of the full CCHS phenotype in regards to respiratory control deficit and ANSD features.17
The variable PHOX2B gene mutations result in variable levels of physiologic dysfunction at the cellular level including (1) altered regulation of genes involved with ANS development such as dopamine beta hydroxylase (DBH),10 (2) altered localization of PHOX2B protein such that it is found in the cytoplasm instead of nucleus of the cell, and (3) altered/absent DNA binding of the PHOX2B protein due to aggregate formation which also interferes with the activity of the normal PHOX2B protein.10,18 These different mechanisms of cellular dysfunction will ultimately determine the severity of each patient’s clinical features, thereby leading to the strong interest in clarifying PHOX2B-genotype/CCHS-phenotype associations.
Early MRI studies and autopsies were unremarkable in individuals with CCHS. More recent investigations utilizing functional MRI (fMRI) and diffusion tensor imaging (DTI) in a small cohort with suspected CCHS (PHOX2B genetic testing was not performed in all subjects) found brainstem changes in areas known to mediate central chemosensitivity.19 The neuroanatomic defects in CCHS are likely the result of focal loss of PHOX2B expression along with consequences of recurrent hypoxemia and/or hypercarbia. Based on rodent studies and fMRI in humans, the following regions pertinent to respiratory control show PHOX2B expression in the pons and medulla of the brainstem: locus coeruleus, dorsal respiratory group, nucleus ambiguus, and parafacial respiratory group, among other areas. Physiologic evidence suggests that the respiratory failure in these children is mostly based on defects in central mechanisms, but peripheral mechanisms (mainly carotid bodies) also are important.19
The majority of PHOX2B mutations causing CCHS are de novo events with a subset of cases that are inherited in an autosomal dominant manner6 from an affected parent (typically, genotype 20/25) or parent with somatic mosaicism,20 even very low-level mosaicism (5%)21 which may note be identified with commonly available sequencing tests.20,21 Germline mutation has been described in one case report,22 emphasizing the importance of genetic counselling and prenatal testing.
Overall, these findings emphasize the importance of requesting PHOX2B testing, with particular attention being given to the testing method. Table 37.1 provides indications and limitations of the clinically available genetic testing methods. A three-step process is generally recommended that includes fragment analysis (screening test), sequencing, and multiplex ligation-dependent probe amplification (MLPA). Since the most common genotypes are PARMs, the majority of mutations (≈90%) will be identified using fragment analysis. Fragment analysis should be performed using assays that amplify the GC-rich areas in order to detect polyalanine regions expanded to 30–33 repeats.23 Sequencing is the most commonly available testing method and will identify all NPARMs and the majority of PARMs. With normal results from both the screening and sequencing tests and with continued clinical suspicion of CCHS, MLPA should be performed to identify large deletions or duplications involving exon 3 of the PHOX2B gene.
Table 37.1
Clinically Available PHOX2B Testing for Individuals with Suspected Congenital Central Hypoventilation Syndrome (CCHS) and Parents of Affected Children
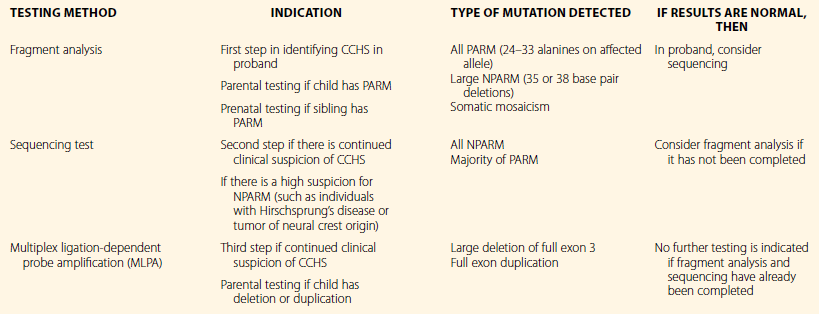
PARM, polyalanine expansion repeat mutation; NPARM, non-polyalanine expansion repeat mutation.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


