Congenital Atresias, Stenosis, and Webs
Andrea Winthrop and Jill Whitehouse
Congenital intestinal atresias, stenosis, and webs typically present early in neonatal life due to the associated obstructive symptoms. The reported incidence varies from 1 to 2 in 5000 births in the United States. These malformations can occur throughout the intestinal tract, but are most commonly found in the duodenum (50%) and the jejunum or ileum (35–40%). A colonic location is identified in 5% to 10% of atresias, and rarely, pyloric or gastric lesions are present (< 1%).1
 PATHOPHYSIOLOGY AND GENETICS
PATHOPHYSIOLOGY AND GENETICS
The etiology most commonly accepted for jejunoileal and colonic atresias involves intrauterine mesenteric ischemia from a variety of vascular accidents, encompassing umbilical cord infarcts, volvulus, internal hernias, fascial defects, intussusception, and intestinal perforation. The affected segment is then reabsorbed. In contrast, duodenal atresia is attributed to a failure of the recanalization process. Genetic links and familial patterns of intestinal atresias (mostly autosomal recessive) are also well described, although the specific genes or mutations involved have yet to be identified. Duodenal atresia is associated with anomalies including trisomy 21, congenital heart disease, malrotation, esophageal atresia, imperforate anus, genitourinary disorders, and gallbladder agenesis. Recently, an autosomal recessive syndrome comprising neonatal diabetes mellitus, intestinal atresias, and gallbladder agenesis has been described.2 An autosomal recessive genetic disorder called Herlitz syndrome is linked to some cases of pyloric atresia. Patients with this syndrome suffer from epidermolysis bullosa lethalis, which results in severe vesiculobullous lesions after minimal skin friction.
 CLINICAL FEATURES, DIAGNOSIS, AND TREATMENT
CLINICAL FEATURES, DIAGNOSIS, AND TREATMENT
The majority of infants with intestinal atresia present as newborns with signs and symptoms of intestinal obstruction (see Chapter 389), including vomiting (which may be bilious), varying degrees of abdominal distension, and often failure to pass meconium. Antenatal diagnosis has been reported, facilitating prompt postnatal diagnosis and management but without proven impact on long-term outcome.3
Antral and Pyloric Atresia and Webs
Antral or pyloric atresias and webs are rare lesions that present typically with nonbilious emesis and failure to thrive.4 Unlike acquired hypertrophic pyloric stenosis, these neonates present with polyhydramnios in utero and an enlarged stomach at the time of birth. Partial antral and duodenal webs can also present with chronic vomiting, often commencing upon the advent of solid feedings, due to intermittent or partial obstruction.5 The most common abnormality is a pyloric web, which is a membrane that can become redundant over time and prolapse into the pyloric channel, creating a classic “wind-sock” finding. Diagnosis of pyloric atresia typically requires an upper GI contrast study and/or endoscopy. Following adequate fluid resuscitation and gastric decompression, treatment includes laparotomy followed by pyloroplasty, gastroduodenostomy, or primary anastomosis, depending on the type and severity of the lesion. Endoscopic laser treatment of an antral web has been reported.6
Duodenal Atresia and Webs
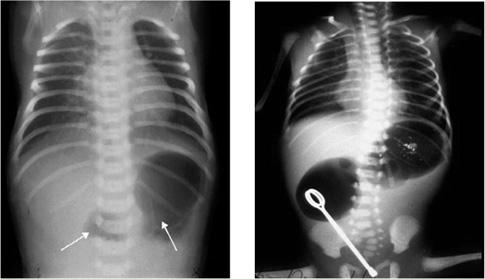
Duodenal atresia and webs account for half of all intestinal atresias. In contrast to jejunoileal atresias, duodenal lesions are more commonly associated with other anomalies, occurring in 50% to 80% of cases.1 As described for pyloric lesions, there are 3 types of atresias in the duodenum, with equal distribution between the incidence of webs and atresias. The usually presentation is bilious vomiting in the newborn. Incomplete obstruction from a fenestrated web (with or without a windsock) or stenosis may have a delayed presentation. Prenatal imaging along with polyhydramnios may suggest the diagnosis, but it is confirmed with an X-ray at birth revealing the classic double-bubble sign (air in both duodenal bulb and stomach as shown in Fig. 399-1). With complete duodenal obstruction, no distal gas will be seen and no further imaging is warranted. If an incomplete obstruction is present, distal gas may confuse the diagnosis with malrotation, and thus an upper GI contrast study should be performed. If malrotation is ruled out, other associated anomalies can be evaluated before proceeding to the operating room. The vast majority of duodenal malformations occur distal to the ampulla of Vater, and hence, great care must be taken to avoid injury to the biliary system intraoperatively. The surgical procedure of choice is a Kimura diamond duodenoduodenostomy. Duodenal webs are managed with incision or partial excision through a duodenotomy. Postoperatively, naso-gastric decompression is sufficient while awaiting bowel function return and normal gastric emptying.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


