Clinical Approach to Neurologic Disease
Maitreyi Mazumdar
HOW TO USE THIS SECTION
This introductory section on pediatric neurology provides a clinically oriented guide to the evaluation of a child with suspected neurologic dysfunction. First we review a basic clinical approach and then we tackle specific neurologic presentations. We attempt to provide a broad framework that will help clinicians begin a thoughtful, focused workup of a child suspected of having a neurologic problem. After review of a basic approach, our discussions are organized around common neurologic presentations and physical findings. This section is not intended to be a comprehensive review of all neurologic disorders; instead, it should be used a starting point. The later sections of this chapter provide further detail about the pathophysiology, diagnoses, and management of these disorders. For reviews of the neurologic examination of infants and children, we refer the reader to Chapter 44 and the monographs and chapters by Dodge and Volpe referenced at the end of this chapter.1,2
THE CLINICAL APPROACH TO A CHILD WITH SUSPECTED NEUROLOGIC DYSFUNCTION
Diseases of the nervous system have a profound impact on the lives of infants, children, and their families. These disorders include epilepsy, cerebral palsy, mental retardation, learning disabilities, complex metabolic diseases, nerve and muscle diseases, and a host of other highly challenging conditions. The clinician approaching an infant or child suspected of having a neurologic disorder faces an imperative to provide an accurate diagnosis. The diagnosis will direct therapy and help the patient and family prepare for possible disability, as well as inform decision making regarding future children.
Medical students, residents, and experienced clinicians often find pediatric neurology inaccessible. Many are intimidated by the intricacy of neuroanatomy, the complexity of biochemical pathways, and the subtlety of a neurologic examination. This situation is compounded by the use of advanced diagnostic technologies such as electroencephalogram (EEG) and sophisticated brain imaging techniques that receive little emphasis in medical school training.
We believe clinicians can overcome many of the difficulties they experience in this area by adhering to the basic principles of clinical medicine and neurology.3 We will review these principles and the important contributions each makes to the diagnostic process.
1. Recognition of impaired neurologic function through careful history and physical examination
2. Identification of the specific part of the nervous system that has been affected (the section “Localization”)
3. Definition of the most likely etiologies, using mode and speed of onset, evolution of illness, and involvement of other organ systems, as well as relevant past and family histories (the section “Differential Diagnosis”)
4. Use of laboratory tests and other diagnostic technologies to determine which of the different possible etiologies is present
5. Assessment of the degree of disability
6. Initiation of therapy, if available, after weighing potential risks and benefits
 HISTORY
HISTORY
Eliciting a careful history remains the cornerstone of the evaluation of children suspected of having neurologic disease. The purpose of the history is to define the nature and temporal profile of the neurologic complaint. The history should be obtained from the patient and parents; observations of teachers or others may also be important. The examiner should take the time to ask all questions needed to understand the details of the problem, including time of onset, exacerbating and alleviating factors, antecedent illnesses, and prenatal and perinatal conditions. If the complaints are paroxysmal and stereotyped, it is frequently useful to ask the child to describe the most recent episode in detail. The symptoms must be defined clearly, as terms have different meanings to different people. The word dizzy, for example, could mean light-headedness or vertigo. Light-headedness would be more suggestive of syncope, whereas vertigo might be caused by vertebrobasilar insufficiency.
Children presenting with headaches, abdominal pain, or reluctance to attend school may well have associated neurologic disturbances. Contributing factors may include previously unrecognized mental retardation, specific learning disabilities, and depression. Less frequently, such complaints may be the harbingers of more serious neurologic illness, such as hydrocephalus or encephalitis.
When evaluating a newborn, the history includes detailed questions about the pregnancy, including in vitro fertilization, maternal drug use, illness during pregnancy, and the presence and quality of fetal movements. The method of delivery and delivery complications should be ascertained as well as birth weight and head circumference. In the evaluation of children, the history includes a careful assessment of development because failure to reach developmental milestones or subtle developmental regression may be early signs of neurologic disease. For example, a young child with gradual loss of the ability to use and understand spoken language may be found to have Landau-Kleffner syndrome, a rare neurologic disorder associated with frequent seizures. Similarly, a young boy with X-linked adrenoleukodystrophy, a uniformly fatal disease characterized by progressive spasticity, obtundation, and adrenal insufficiency, will often present first with subtle behavioral change and developmental regression.
Recognition of similar complaints in other family members is relevant because many neurologic disorders are genetically determined. At minimum, a family history should include an explicit statement of the age and health status of all siblings, parents, parents’ siblings, and grandparents. Particular note should be made of any family members with mental retardation or deaths at an early age. Psychiatric histories of family members should not be neglected, as older family members classified as having schizophrenia or alcoholism may have had neurodegenerative disorders such as Huntington disease.
 GENERAL MEDICAL EXAMINATION
GENERAL MEDICAL EXAMINATION
The general medical examination is essential to a neurologic assessment and frequently provides clues to neurologic disease. The head circumference of all infants and young children should be recorded. Large or small head size often implies intracranial abnormalities. Inspection of the hair, nails, and skin may provide an immediate diagnosis (eg, a neurocutaneous syndrome such as neurofibromatosis-1). The Wood’s lamp is useful in bringing out hypopigmented skin lesions characteristic of tuberous sclerosis; this test should be performed on all infants presenting with infantile spasms. Examination of the lungs and the breathing pattern may uncover disturbances in respiration that reflect brainstem disease or neuromuscular abnormalities. Similarly, the presence of a mass in the abdomen or hepatosplenomegaly may indicate a diffuse process within the central nervous system, such as a storage disorder or cancer with metastasis. Asymmetries of the limbs may suggest atrophy or hemihypertrophy and provide clues to underlying disease.
 NEUROLOGIC EXAMINATION
NEUROLOGIC EXAMINATION
The neurologic examination itself should be as complete as possible and largely guided by historical data. The neurologic examination is traditionally compartmentalized: mental status, craniospinal examination, cranial nerve examination, motor and sensory examination, evaluation of coordination, and assessment of autonomic function. In infants and young children, the examination is best conducted without following a rigid format. Attempts at an “adult-type” examination may lead to crying and fussing and provide only limited information. Children should be watched in their spontaneous activity. Playing or otherwise interacting with the child is the best way to assess cognition, social adaptation, and language. By asking the child to jump, run, write, and use toys, the clinician can assess gross and fine motor function. With some imagination, sensitivity and patience, the experienced clinician can expect to complete the neurologic exam satisfactorily in all children.
Abnormal neurologic findings come in the form of abnormal behavior, developmental regression, impaired posture or gait, difficulty with movements of the face or extremities, and sensory disturbances, including pain.4 Inexperience in examining a child often results in overlooking a neurologic deficit and therefore missing a diagnosis. For instance, mild chorea may appear as normal fidgetiness in a child with Sydenham chorea, a defining feature of acute rheumatic fever. A child with a peripheral neuropathy may come to medical attention because of ataxia; without a careful sensory examination including assessment of proprioception, the neuropathy may be missed. Repeated examinations may be necessary to establish the fundamental clinical findings and ascertain the course of the illness; there is a saying that in a difficult neurologic case, a second examination is the most helpful diagnostic test.3
 LOCALIZATION
LOCALIZATION
After neurologic dysfunction has been recognized through history and physical examination, the next step for the clinician is to identify what part of the nervous system is involved. This process, called localization, requires a basic understanding of neuroanatomy. The first distinction the general practitioner needs to make is whether the process (or “lesion”) involves the central nervous system (brain and spinal cord) or the peripheral nervous system (anterior horn cell, peripheral nerve, neuromuscular junction, and muscle). This distinction is very important as it directs further diagnostic workup. Localization constitutes the foundation of clinical neurology and can take years of diligent study to master. Despite its complexities, some basic principles of localization should be familiar to all physicians and are described below and summarized in Table 547-1. Much of the following is adapted from the classic textbook on localization by Brazis, Madseu, and Biller.5
Localization tends to be most precise when the lesion affects the peripheral nervous system. At the simplest level, injury to a muscle impairs the movement mediated by that muscle. Diseases that affect the neuromuscular junction cause diffuse, fluctuating weakness that often involves muscles of eye movement. Injury to a peripheral nerve causes weakness of the muscles innervated by that nerve and sensory loss in its cutaneous distribution. Lesions of the anterior horn cell present with weakness and later with fasciculations.
In the central nervous system, lesions in the spinal cord below the cervical level cause weakness of one or both legs and sensory loss often characterized by a horizontal level in the trunk. Lesions in the cervical cord or brainstem typically cause weakness or sensory loss on one or both sides of the body. Lesions of the lower pons give rise to gaze palsies and/or peripheral facial weakness. The localization of lesions in the cranial nerves is fairly straightforward, because these lesions cause deficits in the particular functions performed by cranial nerves, such as eye or facial movements.
As we ascend the neuroaxis, localization becomes less precise. Lesions in the cerebellum may cause ataxia. Lesions in the thalamus often cause sensory loss or memory loss. Lesions in the hemispheric white matter may give rise to weakness or visual field defects. Finally, lesions in the cortex manifest themselves by an array of motor, sensory, or behavioral findings that vary according to the area that has been injured. In young infants and children, lesions in the cortex may present as failure to reach developmental milestones or as developmental delay.
 DIFFERENTIAL DIAGNOSIS
DIFFERENTIAL DIAGNOSIS
Because different disease processes affect the same parts of the nervous system, they may often present with similar signs and symptoms. For example, cerebellar ataxia may be the result of a genetic defect, a viral illness, or neo-plastic disease. Similarly, infantile spasms are found in infants with tuberous sclerosis and hypoxic-ischemic encephalopathy as well as variety of other disorders. To form a differential diagnosis, the clinician must be aware of the clinical detail of many disease entities, including their mode of onset, course, and natural history. Many of these details are well known; they will be reviewed later in this chapter and form the substance of following chapters.
Although pediatric neurology seems to many to be a collection of rare and unusual diseases with little relevance to a general pediatric practice, it is important to remember that even in here, unusual presentations of common diseases are more frequent than common presentations of rare diseases. For example, a previously healthy child with an abrupt onset of ataxia and abnormal eye movements is more likely to be suffering from drug ingestion or a viral infection than a rare neurodegenerative disease. In such a case, although the clinician should be aware of neurodegenerative disorders when forming a differential diagnosis, the search for a rare disease should not drive the diagnostic process.
Table 547-1. Distinguishing Features That Aid in Localization of Motor System Disorders
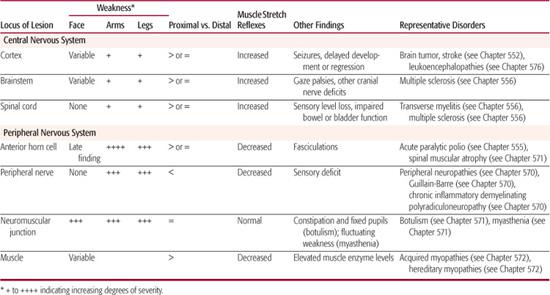
In children especially, every attempt should be made to look for a parsimonious diagnosis, or the simplest explanation of the patient’s signs and symptoms. For example, a child with ataxia, restricted eye movements (ophthalmoplegia), and areflexia is more likely to have the Miller-Fisher variant of Guillain-Barré syndrome than three separate, yet simultaneous, neurologic processes.
 LABORATORY TESTING AND DIAGNOSTICS
LABORATORY TESTING AND DIAGNOSTICS
Ancillary studies, such as laboratory testing, imaging, and neurophysiologic testing, may be planned intelligently only on the basis of clinical information. A “gun-shot” approach to testing, in which a broad array of tests is ordered at the outset, may expose the young child to unnecessary pain and risk and often derails the clinical process by uncovering incidental findings that require further investigation. The approach we suggest is to use ancillary testing initially to exclude items on the differential diagnosis that pose immediate harm to the patient. Once those items have been excluded, a thoughtful, directed workup may proceed at a slower pace, with emphasis placed on noninvasive procedures. We briefly describe some diagnostic tests commonly used in the evaluation of child with neurologic dysfunction.
Computed Tomography (CT)
Computed tomography (CT) scanning revolutionized the practice of neurology and neurosurgery by providing a noninvasive technique to show the intracranial and intraspinal structures. Conventional x-radiation is attenuated as it passes through the skull, cerebrospinal fluid, cerebral gray and white matter, and blood vessels. The intensity of the exiting radiation is measured and the data are integrated by computer. One can see hemorrhage, edematous brain, abscess, tumor tissue, calcifications, and the position of the ventricles and midline structures.
By its nature, CT involves larger radiation doses than the more common, conventional x-ray imaging procedures. Accumulating evidence from epidemiologic studies suggests that the organ doses corresponding to a common CT scan result in increased risk of cancer in children.6 For this reason, the use of CT scans should be considered carefully. The head CT is most appropriate in the evaluation of head trauma and the assessment of ventricular size in suspected cases of shunt malfunction and hydrocephalus. Even in the latter case, there are efforts to replace CT with limited magnetic resonance imaging (MRI) because of concerns about radiation exposure.
Magnetic Resonance Imaging (MRI)
Magnetic resonance imaging (MRI) produces detailed images using the body’s natural magnetic properties, specifically the hydrogen proton because of its abundance in water and fat. The basic elements of MRI scanning are discussed in detail in (see Chapter 552). Areas poorly seen on computed tomography (CT) scan (eg, posterior fossa and spinal cord) can be defined clearly by MR imaging. With the exception of detecting calcification/mineralization, MR imaging surpasses that possible by CT.
The clinical situation guides the radiologist’s choice of sequences, and thus clinical information should be communicated clearly when ordering an MRI. For example, a teenager with acute onset of arm and leg weakness after being tackled on the football field has a history that suggests acute stroke secondary to traumatic arterial dissection. The MRI should include specialized sequences such as diffusion-weighted-imaging (DWI), a sequence helpful in identifying acute ischemia. In addition, magnetic resonance angiography (MRA) provides visualization of the main intracranial arteries and can reliably detect vascular lesions and stenosis. A child with suspected infection or brain tumor might receive gadolinium contrast during the MRI, as this agent permits even sharper definition and highlights areas where the blood-brain barrier has been disrupted.
The degree of cooperation required to perform MR imaging limits its use in young children. Children often need to be sedated in order for the test to be properly performed. The main risks of MR imaging are dislodgement of metal clips and other ferromagnetic objects.
Cerebral Angiography
The use of conventional angiography has decreased remarkably due to the increased availability of CT and MRI. Its use is limited to diagnosis of aneurysms, vascular malformations, and arterial dissections. Following local anesthesia, a needle is placed in the femoral or brachial artery. A cannula is then threaded through the needle, along the aorta and the arterial branches to be visualized. In this way a contrast agent can be injected into the carotid and vertebral systems and to their extents in the neck and cranial cavity. Through the use of digital subtraction—a digital computer process to produce images—the vessels can be visualized with smaller catheters and smaller amounts of dye than was previously possible. Because of the risks of vascular spasm and of embolization of the artery from clot formation at the tip of the catheter, conventional angiography should not be undertaken unless it is absolutely necessary. The applications of cerebral angiography are discussed further in Chapter 552.
Electroencephalography (EEG)
Electroencephalography (EEG) continues to be an essential part of the study of children with seizures and those suspected of having seizures. EEG is also used in evaluation of the effects on the brain of many metabolic diseases, in the study of sleep, and in the operating room to monitor cerebral activity in anesthetized patients. EEG records spontaneous electrical activity generated in the brain. Metal electrodes are coated with conductive paste and placed on the scalp. Differences in voltage potential between electrodes are amplified and displayed on a screen. Digital systems permit reconstruction of the EEG with any desired derivation or format, and permit data manipulation for added analysis. Patients should be studied in the waking and sleeping states; activation procedures such as photic stimulation and hyperventilation are used to elicit epileptiform tendencies.7 The classification of epileptic seizures involves both clinical and electroencephalographic criteria, and is described further in Chapter 557.
Electromyography and Nerve Conduction Studies (EMG/NCV)
Electromyography (EMG) and nerve conduction studies (NCV) assess abnormalities in the peripheral nervous system. EMG is used to detect abnormal muscle electrical activity that can occur in many diseases, including muscular dystrophy and myositis. It is most helpful in distinguishing disorders of muscle from disorders of nerves. Nerve conduction studies involve stimulation of motor or sensory nerves and recording of the elicited responses. The test is used to identify nerve damage or destruction, such as that found in demyelinating diseases and hereditary neuropathies. EMG and nerve conduction studies are discussed in greater detail in Chapter 568.
 EVALUATION OF DISABILITY
EVALUATION OF DISABILITY
Once a diagnosis is established, the clinician’s next step is to determine the level of disability, if any, that is present. Two children may suffer from the same disease process, yet have very different levels of function. Chronic headache, for example, can lead one child to miss weeks of school and withdraw from social activities, whereas another child with the same disorder functions well with few limitations. The clinician must be sensitive to the individual child’s experience. The teenager who is missing weeks of school because of migraines is at high risk for developing comorbidities, such as a major depressive disorder. This patient requires more intensive intervention, including counseling and pain management specialists.
 THERAPY
THERAPY
In the recent past, neurologic diseases in children were thought to be untreatable and the clinical method described above was considered to be little more than an intellectual exercise. Through advances in neuroscience, immunology, pharmacology, microbiology, and other fields, such is no longer the case. Specific therapies are now available for a growing number of diseases. The use of intravenous immunoglobulin (IVIG) for Guillain-Barre, for example, has completely changed the expected outcome for children with this disorder. Prior to the use of IVIG, many of these children died from respiratory failure; now these children regain full strength within weeks. Similarly, advances in surgery and chemotherapy have made childhood brain cancer a survivable disease for many children.
COMMON SIGNS AND SYMPTOMS OF NEUROLOGIC DISEASE
The goal of the following sections is to use the clinical method described above to frame the evaluation of common neurologic problems. In some of the examples, we focus mostly on history; in others, we focus on using the neurologic examination to guide localization; in others, we expand upon a differential diagnosis. We intend that these examples provide a starting point for the clinician. For a more comprehensive review, we reference the later chapters in this book that discuss these issues.
 WEAKNESS
WEAKNESS
A systematic approach is necessary when evaluating a weak child, and the first question is whether the weakness is in fact due to a problem involving the nervous system. Many processes outside of the nervous system, such as febrile illness, chronic infections, electrolyte imbalances, and endocrinopathies are associated with weakness. These disorders most often lead to generalized symptoms and a lack of objective findings on neurologic examination.
The history should focus on the time course of the weakness, as this information provides clues to the etiology. Static weakness suggests a discrete injury, such as an infarction. Rapidly evolving weakness is most often caused by a Guillain-Barré syndrome, an acute demyelinating polyneuropathy. Episodic weakness suggests a paroxysmal cause, such as complicated migraine or epilepsy. Weakness that progresses in a slow and indolent fashion poses the greater difficulty in diagnosis, as it can suggest a neurodegenerative process or slowly growing tumor.
The primary purpose of the examination in these cases is to localize the anatomic site of the lesion causing weakness. Distinguishing features of the examination that aid in localization are described below and summarized in Table 547-1.2,8 Disorders related to central causes (ie, in brain or spinal cord) are characterized by weakness of the limbs, preserved or hyperactive deep tendon reflexes, and an absence of fasciculations. Other indications of cerebral disease include seizures and diffuse developmental delay. Central causes are as diverse as congenital malformation, hydrocephalus, transverse myelitis, disorders of neural tube development, and mass lesions such as tumors or empyemas. Brain or spinal imaging with magnetic resonance imaging (MRI) is likely to be helpful.
Diseases of the anterior horn cell are characterized by generalized weakness, absent reflexes, fasciculations, intact mental status, and normal sensation. Before the widespread use of vaccine, anterior horn cell disease in children was most often due to poliovirus infection. Acute paralytic poliomyelitis (Chapter 555) usually presents with asymmetric flaccid weakness and is usually preceded by nonspecific symptoms such as low-grade fever and diarrhea. Spinal muscular atrophy (Chapter 571) is an autosomal recessive disorder of the anterior horn cells. The most common form presents within the first years of life and is marked by progressive weakness with absent reflexes, tongue fasciculations, and eventually respiratory failure. Electromyography (EMG) and genetic tests confirm the diagnosis.
Diseases of the peripheral nerve cause generalized weakness and decreased reflexes sometimes accompanied by sensory disturbances. Guillain-Barre syndrome is an inflammatory disease that produces rapidly progressive symmetric ascending flaccid weakness with depressed reflexes and variable sensory findings (Chapter 570). Inherited neuropathies can also produce weakness, although the time course is usually much more gradual. Motor neuropathies such as Charcot-Marie-Tooth disease are reviewed in Chapter 570. Nerve conduction studies are commonly used to confirm the diagnosis of a motor neuropathy; an initial serum creatinine kinase may distinguish nerve-mediated weakness from muscle disease.
Diseases of the neuromuscular junction are characterized by weakness involving the face, eyelids, and extraocular muscles. Physical exam reveals normal deep tendon reflexes and sensory function. In infants, botulism is the prototypical disease of the neuromuscular junction. An infant affected by botulism will have a relatively acute onset of weakness preceded by difficulty feeding and constipation (Chapter 571). Myasthenia gravis, an autoimmune disorder most often caused by antibodies to the acetylcholine receptor in the motor end plate, causes fluctuating weakness that is more prominent with exercise and improves with rest (Chapter 571). EMG and antibody testing are used to confirm these diagnoses as well.
Muscle diseases are typified by weakness that is greater in the proximal limbs. A child with an acquired inflammatory myopathy, such as that seen in viral myositis, might initially have a low-grade fever and then report difficulty getting up from a chair. Duchenne muscular dystrophy is characterized by decreasing muscle mass and progressive loss of muscle function in male children. Early signs may include difficulties standing up and walking on stairs. The weakness progresses and the young boy eventually needs a wheelchair. Infants with congenital myopathies will present with generalized weakness, poor muscle bulk, and dysmorphic features that may be secondary to the weakness. Myopathies are often associated with other organ systems, such as the heart and skin. Initial evaluation usually includes serum creatinine kinase, an enzyme found primarily in muscle. Elevated levels of creatinine kinase suggest significant muscle injury such as that found in myopathy. Myopathies are reviewed in Chapter 572.
 ATAXIA
ATAXIA
Ataxia is defined as impaired coordination of movement and balance. The incoordination can be caused by processes as diverse as drug ingestion, varicella infection, brain tumors, and hereditary neurodegenerative disease. The pathology can be located at any level of the nervous system, from cerebral cortex to muscle. Ataxia is particularly frightening to children and parents, because it represents a loss of physical control and raises the possibility of some very debilitating diseases. As is usually true, a careful history and physical examination, followed by focused radiologic and laboratory assessments, can lead to proper diagnosis and timely intervention.
The time course of the symptoms usually provides the greatest information about ataxia.9-11 When taking the history, the clinician should determine whether the onset was acute (suggesting viral infection, drug ingestion, or the Miller-Fisher variant of Guillain-Barré syndrome), subacute over weeks (brain tumors, nutritional deficiencies, or paraneoplastic syndromes), or chronic over months to years (cerebellar degeneration or hereditary metabolic diseases). Other elements of the history should include whether the child has had a previous episode, whether there are medications in the house, and a full medical and family history, including that of migraines and epilepsy as these can also present with ataxia.
The general medical examination should include optic funduscopy to look for signs of increased intracranial pressure. The skin should be evaluated for viral exanthems and crusted lesions typical of varicella. Pharyngitis, cervical adenitis, and splenomegaly point toward other infectious etiologies such as mononucleosis. Cardiac assessment may reveal murmurs or arrhythmias that could lead to embolic stroke. Pes cavus and scoliosis may be apparent in a child with Friedreich ataxia, an autosomal recessive disorder. Oculocutaneous telangiectasias are often visible in a child with ataxia telangiectasia, also an autosomal recessive disorder characterized by progressive truncal ataxia and recurrent sinus infections.
The neurologic examination should attempt to provide information that aids localization, focusing on the cerebellum and its major input systems from the frontal lobe and posterior columns of the spinal cord. When an abnormality occurs in the vermis of the cerebellum, the child cannot sit still but constantly moves the body to and fro. In contrast, disturbances of the cerebellar hemispheres cause dysmetria and hypotonia. Bifrontal lobe disease may produce signs indistinguishable from those of cerebellar disease. Other features of cerebellar disease are a characteristic scanning speech and intention tremor. Loss of sensory input to the cerebellum because of peripheral nerve or posterior column disease causes a careful and hesitant gait that is worse when the eyes are closed. Children with weakness often stagger about, which can be mistaken for ataxia. Muscle strength must be specifically assessed.
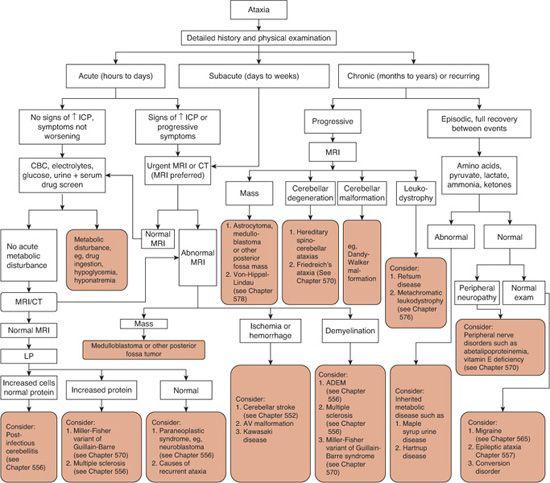
The laboratory and radiologic workup for ataxia is outlined in Figure 547-1. The slow evolution of ataxia in a previously healthy child warrants rapid evaluation, as the most immediate concern is brain tumor; brain imaging should be performed without delay. The work-up for acute ataxia should include a complete blood count, measurement of electrolytes and glucose, toxicology screening of blood and urine, brain imaging, and lumbar puncture. If magnetic resonance imaging (MRI) or computed tomography (CT) demonstrates a mass lesion, hydrocephalus, or intracranial abnormality, cerebrospinal fluid (CSF) evaluation may be deferred. The presence of cells in the CSF may indicate infection, and elevated CSF protein levels are associated with Guillain-Barré syndrome and multiple sclerosis.
The workup for children with chronic or recurring ataxia is guided by the history and physical examination. Every child with chronic, progressive ataxia should undergo imaging, preferably MRI. Other structural abnormalities that could be found by MRI are cerebellar malformations, although these are most often found in children with other developmental abnormalities.
Special attention needs to be paid to a sensory examination in children with chronic ataxia. Children with sensory neuropathies such as those found in abetalipoproteinemia or vitamin E deficiency will present with difficulty walking and incoordination due to disrupted sensory input to the cerebellum. Children with Friedreich ataxia have degeneration of the posterior columns of the spinal cord in addition to degeneration of other parts of the brain and cerebellum.
Migraine, seizure, and some rare metabolic disorders may manifest with acute intermittent episodes of ataxia. Evaluation is again based on the history, but may include electroencephalogram (EEG) and electromyography (EMG). If metabolic disease is suspected, evaluation of amino acids, acid/base balance, lac-tate, pyruvate, ammonia, and ketones may be helpful. If the workup fails to disclose a diagnosis, referral for a detailed neurologic and genetic evaluation is warranted.
 SEIZURES
SEIZURES
Seizures are among the most common symptoms of disturbed brain function and a cardinal manifestation of neurologic disease. Physiologically, epileptic seizures are caused by an electrical discharge from a group of excitable neurons in any part of the cerebral cortex. This abnormal discharge can be the result of many different causes, ranging from a benign developmental predisposition to a fulminant central nervous system infection. History and physical examination will be able to differentiate the child needing immediate intervention from one whose work-up can proceed at a slower pace. An overview of an approach to the child with suspected seizures is presented in Figure 547-2.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


