16.3 Cancers
Incidence and distribution of childhood cancers
Approximately 1 in 600 children will be diagnosed with cancer before the age of 15 years, and the incidence has slowly increased since the 1970s. The distribution of cancer types in children aged 0–14 years is shown in Table 16.3.1. The incidence varies by sex and ethnic origin, and the types of tumour also vary by age.
Table 16.3.1 Frequency of malignancy in childhood
| Malignant disease | Frequency (%) |
|---|---|
| Leukaemia | 35 |
| Primary central nervous system tumours | 20 |
| Lymphoma: non-Hodgkin and Hodgkin | 10 |
| Wilms’ tumour | 6–8 |
| Neuroblastoma | 6–8 |
| Rhabdomyosarcoma, soft tissue sarcoma | 5 |
| Sarcoma of bone: Ewing and osteosarcoma | 4 |
| Histiocytosis | 5 |
| Teratoma | 2 |
| Retinoblastoma | 1 |
| Hepatic | 1 |
| Other | 5 |
Aetiology of childhood cancer
When confronted with a diagnosis of childhood cancer, parents often ask: ‘Why did this happen to my child?’ or ‘Did this happen because of something I have done or passed on to my child?’ With the exception of several known predisposing genetic syndromes (Table 16.3.2), the proportion of paediatric cancers that have a clearly hereditary component is very small. Similarly, despite extensive epidemiological studies, few environmental agents have been linked consistently with childhood malignancy.
Table 16.3.2 Inherited/genetic syndromes associated with increased risk of childhood malignancy
| Cancer | Associated syndrome |
|---|---|
| Leukaemia | Trisomy 21, Bloom syndrome, Fanconi anaemia, ataxia telangiectasia, neurofibromatosis, Kostmann syndrome, Klinefelter syndrome, Li–Fraumeni syndrome, Diamond–Blackfan anaemia, Noonan syndrome |
| Central nervous system tumours | Neurofibromatosis, tuberous sclerosis, Li–Fraumeni syndrome, von Hippel–Lindau syndrome |
| Lymphoma | Immunodeficiency disorders |
| Wilms’ tumour | Denys–Drash syndrome, Beckwith–Weidermann syndrome, WAGR syndrome |
| Rhabdomyosarcoma | Li–Fraumeni syndrome |
WAGR, Wilms’ tumour, aniridia, genitourinary anomalies and mental retardation.
Approach to management of a patient with suspected malignancy
• Diagnosis will be made by a combination of diagnostic tests, radiological imaging and biopsies that varies dependent on the cancer type. Examples of these will be shown later as we discuss specific tumour groups.
• Staging investigations are then needed to document whether the cancer has spread. These tests give important information about survival and allow clinicians to decide on the most suitable clinical trial.
• Toxicity assessment such as echocardiography, glomerular filtration rate and audiology are regularly carried out to ensure that treatment does not affect normal structures such as the heart, kidneys and hearing.
• Treatment usually involves combinations of four common treatment options, although scientists are researching new novel treatments that may improve survival:
Acute leukaemia
Leukaemia can be further classified into distinct categories:
Classification is on the basis of:
• Morphological characteristics – appearance of the blood film, bone marrow aspirate and bone marrow trephine under the microscope
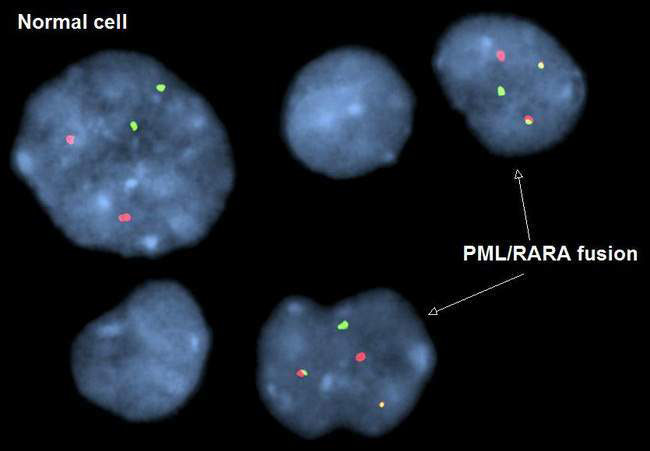
• Cytogenetics – the study of tumour chromosomal material using techniques such as florescence in situ hybridization (FISH) and comparative genomic hybridization (CGH)
• Immunophenotyping – a technique used to identify surface markers and antigens on leukaemia and lymphoma cells to aid diagnosis and classification.
Acute lymphoblastic leukaemia
Investigations
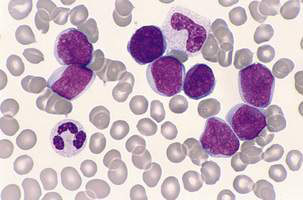
The peripheral blood film can be normal but will usually demonstrate the presence of leukaemic blasts with or without anaemia and thrombocytopenia. The white blood cell count (WCC) is frequently raised at diagnosis (leukocytosis), with a presenting WCC below 10 × 109/L in 25%, 10–50 x 109/l in 50% and above 50 x 109/l in 25% of patients. A bone marrow aspirate and biopsy (trephine) are the ‘gold standard’ diagnostic tests and will show replacement of normal haematopoiesis by leukaemic cells (Fig. 16.3.1).
Classification and prognostic factors
Table 16.3.3 shows prognostic risk factors for ALL. Clinical features such as age and WCC at diagnosis are becoming less significant as protocols stratify treatment based on the response to treatment; for example, reduction of initial blast count following steroid therapy is an important prognostic factor, as is detection of minimal residual disease (MRD) by molecular methods after chemotherapy.
Table 16.3.3 Risk group classification for acute lymphoblastic leukaemia
| Risk group | Clinical features | Molecular/genetic features |
|---|---|---|
| Low risk | Age 2–10 years, WCC < 50 × 109/l | DNA index > 1.16 |
| Not T-cell phenotype | Absence of: | |
| No central nervous system or testicular disease | ||
| Rapid response to induction therapy | ||
| t(12;21) TEL/AML1 | ||
| High risk and very high risk | Induction failure | t(9;22), t(4;11) |
| Age < 12 months | MLL rearrangements | |
| Poor prednisone response | ||
| High MRD levels |
MRD, minimal residual disease; WCC, white blood cell count.

Acute myeloid leukaemia
Classification and prognostic factors
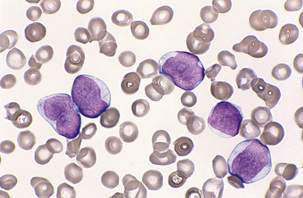
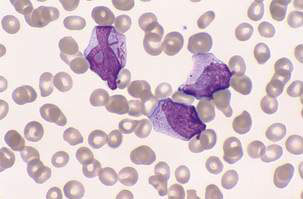
In AML, characteristic morphological features include the presence of Auer rods as well as positive staining for myeloperoxidase and monocyte-associated esterases (Fig. 16.3.2). Classification into one of eight morphological subclasses using the French–American–British (FAB) system is possible.
Treatment

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


