Cancer Genetics and Biology
David Malkin
The unique biological features, the cell of origin, and the response to therapy of childhood cancers make them intriguing models with which to study and understand the process of human carcinogenesis. Some cancers, such as neuroblastoma, appear to arise even before birth, whereas a significant number of others present with a striking family history of cancer or coincide with congenital abnormalities, suggesting an important role for inherited causal genetic factors. 
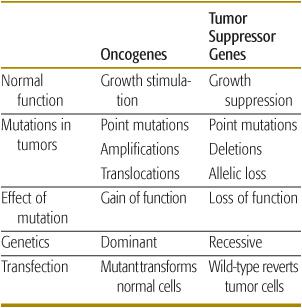
With the advent of technologies that can provide highly sensitive genome-wide molecular analysis, as well as clinical oncologists and geneticists recognizing the value of carefully ascertaining family cancer histories, it is likely that both new cancer predisposition associations and novel cancer genes will continue to be discovered. Importantly, new genetic tests offer opportunities for presymptomatic diagnosis of syndromic associations and a greater role of clinical surveillance by pediatricians and family physicians for early cancer detection in high-risk individuals. Study of both sporadic childhood cancers and familial syndromes associated with childhood malignancies has led to the identification and functional characterization of numerous tumor suppressor genes (TSGs), oncogenes, and DNA repair genes. TSGs normally control cell proliferation by either inhibiting progression through the cell cycle or promoting apoptosis (programmed cell death). Inactivation of both alleles of a TSG by mutation or deletion causes uncontrolled proliferation, thereby contributing to tumorigenesis. In contrast, oncogenes normally potentiate cell proliferation, and mutation of one allele is sufficient to produce uncontrolled cell growth. Inactivation of DNA stability or repair genes permits cells that harbor damaged DNA to divide, potentially leading to malignant transformation of those cells.2 Nonrandom molecular and cytogenetic alterations are frequently observed in most childhood cancers. These markers are unique diagnostic identifiers and frequently provide prognostic value with respect to disease outcome and anticipated response to therapy. Importantly, many of these genetic markers also recapitulate normal developmental processes, and as such offer a window into the biological mechanisms of both carcinogenesis and normal embryologic growth and development (Table 443-1).
THE RETINOBLASTOMA SUSCEPTIBILITY GENE (RB1): PARADIGM TUMOR SUPPRESSOR
Retinoblastoma (RB) is the prototype cancer caused by mutations of a tumor suppressor gene.  This condition occurs in either a sporadic or a hereditary form. Patients with sporadic RB usually have unilateral tumors and are older (median age 22 months) at diagnosis, whereas patients with hereditary RB often develop bilateral or multifocal tumors and are younger (median age 11 months) at diagnosis.4 Based on this inheritance pattern, Knudson proposed the “two-hit hypothesis,” which established the basis for discovery of TSGs.5 According to this hypothesis, the inactivation of both alleles of the causally associated gene is necessary for tumor development. In hereditary tumors, one mutated allele is inherited through the germ line and the second one is inactivated by a somatic event, often a gene deletion. In sporadic tumors, both alleles are inactivated in the somatic cell, leading to their unifocal and later onset.
This condition occurs in either a sporadic or a hereditary form. Patients with sporadic RB usually have unilateral tumors and are older (median age 22 months) at diagnosis, whereas patients with hereditary RB often develop bilateral or multifocal tumors and are younger (median age 11 months) at diagnosis.4 Based on this inheritance pattern, Knudson proposed the “two-hit hypothesis,” which established the basis for discovery of TSGs.5 According to this hypothesis, the inactivation of both alleles of the causally associated gene is necessary for tumor development. In hereditary tumors, one mutated allele is inherited through the germ line and the second one is inactivated by a somatic event, often a gene deletion. In sporadic tumors, both alleles are inactivated in the somatic cell, leading to their unifocal and later onset.
Table 443-1. Properties of Tumor Suppressor Genes and Oncogenes
The RB1 gene was initially localized to chromosome 13 (13q14)6 and was the first TSG to be cloned.7 Loss of function of both alleles of RB1 in tumor specimens8 and the presence of germ-line mutations in hereditary retinoblastoma9 validated Knudson’s two-hit hypothesis. RB1 is mutated or deleted in all retinoblastomas and in a wide spectrum of other tumors.10 It is thought that several genetic mechanisms may be involved in eliminating the second wild-type RB1 allele in an evolving tumor. These include chromosomal duplication or nondisjunction, mitotic recombination, or gene conversion.11-16 The retinoblastoma protein, pRB, serves as the “molecular switch” that controls the passage of cells through division and proliferation.11,12 Loss of this regulatory function allows uncontrolled proliferation, thereby contributing to tumorigenesis. 
The protein pRB plays a central role in the control of cell-cycle regulation, particularly in determining transition from G1 through the S (DNA synthesis) phase in virtually all cell types.21
Although the RB1 gene is expressed in virtually all mammalian tissues, only in the retina is its inactivation sufficient for tumor initiation. Outside the retina, RB1 inactivation is often a rate-limiting step in tumorigenesis generated by multiple genetic events, and these defects—along with the observation that mice lacking the RB1 gene are viable only through day 16 of gestation,28 as well the occurrence of specific organ defects—suggest that the critical role of Rb may be in differentiation of these affected tissues rather than in the control of cell growth per se.29
As the biological mechanisms by which RB1 gene inactivation are better understood,  functional interactions of pRB with its binding partners and other cell-cycle targets will offer opportunities to explore development of novel small-molecule therapies to complement conventional approaches.
functional interactions of pRB with its binding partners and other cell-cycle targets will offer opportunities to explore development of novel small-molecule therapies to complement conventional approaches.
WILMS TUMOR: A COMPLEX TUMOR SUPPRESSOR MODEL
Wilms tumor (WT), or nephroblastoma, is an embryonal malignancy that arises from remnants of immature kidney.32
WT is often associated with specific congenital abnormalities. A genetic predisposition to WT is observed in two distinct syndromes with urogenital system malformations—the WAGR (Wilms tumor, aniridia, genitourinary abnormalities, mental retardation) syndrome and the Denys-Drash syndrome (DDS)37—as well as in Beckwith-Wiedemann syndrome (BWS).38 These congenital disorders have now been linked to abnormalities at specific genetic loci implicated in Wilms tumorigenesis. 
WAGR syndrome is associated with constitutional deletions of chromosome 11q13, which harbors both the aniridia gene Pax639,40 and the WT1 gene.39,41-43 In addition, sporadic and hereditary WT have been described in which WT1 is specifically altered. Tumor-specific deletions at the WT1 locus have been identified and suggest that this allele is specifically inactivated during pathogenesis of some WTs. 
Virtually all patients with DDS carry germ-line WT1 point mutations.48 Germ-line WT1 mutations are also observed in children with hypospadias and other developmental anomalies of the male genitalia.
WT1 is altered in only 10% of sporadic Wilms tumors.45 This implies that other genetic loci are important in its etiology. One such locus also resides on the short arm of chromosome 11, telomeric of WT1, at 11p15. This gene, designated WT2, is associated with BWS. 
Although both tumors represent classic models of the Knudson two-hit hypothesis of tumor development, the spectrum of genetic alterations in Wilms tumor is quite different from that in retinoblastoma. In retinoblastoma, a single gene is involved whose function is mediated through related cell-cycle control pathway genes, whereas a series of distinct genetic events is required for Wilms tumorigenesis. 
TUMORS OF THE PERIPHERAL NERVOUS SYSTEM: NEUROBLASTOMA 
Nonrandom cytogenetic abnormalities are observed in more than 75% of neuroblastomas.57-59 The most common of these is deletion or rearrangement of the short arm of chromosome 1,59 although loss, gain, and rearrangements of chromosomes 10, 11, 14, 17, and 19 have also been reported. 
Two other unique cytogenetic rearrangements are highly characteristic of neuroblastoma.67,68 These contain regions of amplification of the N-myc gene, an oncogene with considerable homology to the cellular proto-oncogene cmyc. N-myc amplification is associated with rapid tumor progression.69 Expression of N-myc is increased in undifferentiated tumor cells compared with much lower (or single-copy) levels in more differentiated cells (ganglioneuroblastoma and ganglioneuroma). Furthermore, a close correlation exists between N-myc amplification and advanced clinical stage.70 Decreased N-myc expression is observed in association with differentiation of neuroblastoma cell.71 This observation formed the basis for current therapeutic trials demonstrating a survival advantage for patients treated with cis-retinoic acid.72
While most neuroblastomas are sporadic in nature, study of rare cases of familial neuroblastoma led to discovery of a familial neuroblastoma gene (REF). Remarkably, this gene (identified using high-throughput genome-wide microarray analysis) is ALK, the tyrosine kinase associated with the t(2;5) translocation characteristic of ana-plastic large-cell lymphoma. Activating mutations of the ALK gene induce proliferation of malignant neuroblasts. Furthermore, somatic mutations of this gene are observed in sporadic neuroblastomas as well, suggesting its importance in the genesis of this tumor. 
The use of tyrosine kinase inhibitors to block the function of aberrant ALK expression offers hopeful opportunities for novel effective therapies for this disease.
CYTOGENETIC TRANSLOCATIONS—THE ROLE OF FUSION PROTEINS IN EWING SARCOMA AND PRIMITIVE NEUROECTODERMAL TUMORS 
A striking distinguishing chromosomal translocation, t(11;22)(q24;q12),84-88 is frequently observed in peripheral primitive neuroectodermal tumor/Ewing sarcoma (PPNET/ES). 
The translocation breakpoint is an in-frame fusion between the 5′ half of the ES gene, EWS, on chromosome 22 and the 3′ half of the human homologue of an ETS transcription family member, Fli-1, on chromosome 11.89-91 Targets of the EWS-Fli-1 transcription factor may include insulin-like growth factor I receptor (IGF-IR),93 which is thought to play a role in the pathogenesis of ES. 
Greater than 90% of the ES family of tumors (ESFTs) carry the EWS-ETS fusion gene, and the specific fusion protein expressed in ESFT has prognostic significance.99,100
While the EWS-Fli-1 fusion protein presents a tantalizing opportunity for development of novel molecular therapies, its sequestration in the nucleus makes it challenging to create effective agents (such as antibodies) that will target the nuclear fraction. Thus, inhibitors of IGF-I signaling appear to hold more promise.
CYTOGENETIC TRANSLOCATIONS OR CHROMOSOMAL LOSS IN RHABDOMYOSARCOMA
Rhabdomyosarcoma (RMS) is the most common soft-tissue sarcoma of childhood, accounting for nearly 10% of all childhood solid tumors.106-108 Multiple histological subtypes exist, including embryonal (ERMS; 63%) and alveolar (ARMS; 19%) morphologies. 
Characteristic genetic lesions have been found in both major RMS subtypes. More than 75% of ARMS demonstrates one of two chromosomal translocations, t(2;13)(q35;q14) or t(1;13)(p36;q14)109-111; these yield fusion between the 5′ DNA-binding region of PAX-3 on chromosome 2 or PAX-7 on chromosome 1, respectively (implicated in neuromuscular development), and the 3′ transactivation domain region of FKHR (FOXO1A) gene (a member of the forkhead family of transcription factors commonly associated with regulation of apoptosis).112
The genetics of ERMS tumors are distinct from ARMS primarily in that they display a specific loss of heterozygosity (LOH) at chromosome 11p15, involving the loss of maternal and duplication of paternal genetic material. Classically associated with this LOH is the overexpression (due to a failure of imprinting) of the insulin-like growth factor II (IGF-II), which may be involved in the tumorigenic cascade that leads to ERMS. 
The LOH at 11p15 occurs by loss of maternal and duplication of paternal chromosomal material.131 Although LOH is normally associated with loss of TSG activity, in this instance, LOH with paternal duplication may result in activation of IGF-2. 
OSTEOSARCOMA: COMPLEX GENETIC MECHANISMS 
The molecular basis of osteosarcoma (OS) is strikingly more complex than that of other bone tumors, such as ES/PPNET. In particular, OS is typically characterized by the presence of complex unbalanced karyotypes in most cases.142 Combined inactivation of the RB1 and p53 tumor suppressor pathways are observed in most OS. 
Common numerical abnormalities in OS include gain of chromosome 1; loss of chromosomes 9, 10, 13 (at the RB1 locus), or 17 (at the p53 locus); and partial or complete loss of the long arm of chromosome 6.147-150 Frequent structural abnormalities include rearrangements of chromosomes 11, 19, and 20.147,148,150-152
Karyotypic complexity may reflect chromosomal fusion-bridge-breakage cycles that occur due to advanced telomere erosion. A potential etiology of this chromosomal instability is telomere dysfunction. 
CANCER PREDISPOSITION SYNDROMES
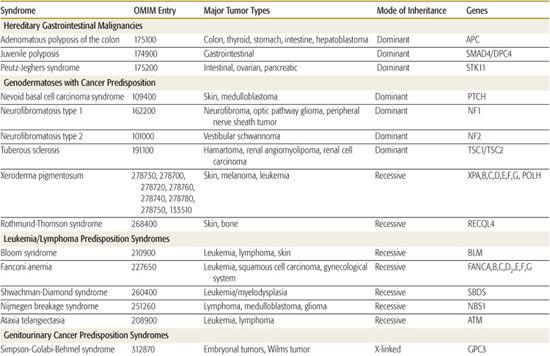
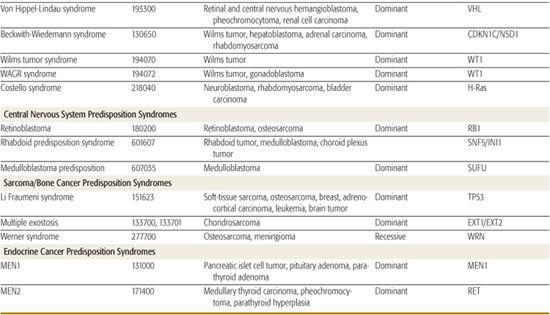
Several hereditary cancer syndromes are associated with the occurrence of childhood and adult-onset neoplasms. This chapter discusses a few to highlight the important molecular basis on which these disorders form (Table 443-2).
 LI-FRAUMENI SYNDROME
LI-FRAUMENI SYNDROME
Li-Fraumeni syndrome (LFS) requires a proband with a sarcoma diagnosed before 45 years of age and a first-degree relative with any cancer under 45 years of age and a first- or second-degree relative with any cancer under 45 years of age or a sarcoma at any age.4 The classical spectrum of tumors that includes soft-tissue sarcomas, osteosarcomas, breast cancer, brain tumors, leukemia, and adrenocortical carcinoma (ACC) has been overwhelmingly substantiated by numerous subsequent studies175; however, other cancers, usually of particularly early age of onset, are also observed.176
Germ-line alterations of the TP53 tumor suppressor gene are associated with LFS.179-181 Carriers are heterozygous for the mutation, and in tumors derived from these individuals, following Knudson’s two-hit model, the second (wild-type) allele is frequently deleted or mutated, leading to functional inactivation.182-184
Between 3% and 10% of children with apparently sporadic rhabdomyosarcoma or osteosarcoma carry germ-line TP53 mutations.186-189 These patients tend to be younger than those who harbor wild-type TP53. More than 75% of children with apparently sporadic adrenocortical carcinoma harbor germ-line TP53 mutations, although in some of these cases, a family cancer history frequently evolves with time.190,191 These observations suggest that germ-line TP53 alterations may be associated with early onset development of the childhood component tumors of LFS.194
While germ-line TP53 mutations establish the baseline risk of tumor development in LFS, a complex interplay of modifying genetic cofactors likely define the specific phenotypes of individual patients.
The primary treatment of patients with LFS is directed to the specific tumors.  Most conventional chemotherapeutic agents—such as the anthracyclines, alkylators, and platinum-based agents—and therapeutic doses of radiation induce cell growth arrest or apoptosis through a p53-mediated signaling pathway. Thus, to modify therapy in the face of mutant p53 is challenging and would severely limit the selection of effective anticancer therapy. For this reason, several approaches to developing wild-type gene replacement therapy or small molecules that enhance expression of wild-type p53 are under way. With the identification of germ-line TP53 mutations in asymptomatic children or young adults, it becomes possible to consider introducing aggressive clinical surveillance protocols to enhance the likelihood of early detection of cancers. While this approach may be clinically appropriate, the genetic testing of children raises many ethical and philosophical concerns that are the focus of intense scrutiny.
Most conventional chemotherapeutic agents—such as the anthracyclines, alkylators, and platinum-based agents—and therapeutic doses of radiation induce cell growth arrest or apoptosis through a p53-mediated signaling pathway. Thus, to modify therapy in the face of mutant p53 is challenging and would severely limit the selection of effective anticancer therapy. For this reason, several approaches to developing wild-type gene replacement therapy or small molecules that enhance expression of wild-type p53 are under way. With the identification of germ-line TP53 mutations in asymptomatic children or young adults, it becomes possible to consider introducing aggressive clinical surveillance protocols to enhance the likelihood of early detection of cancers. While this approach may be clinically appropriate, the genetic testing of children raises many ethical and philosophical concerns that are the focus of intense scrutiny.
 BECKWITH-WIEDEMANN SYNDROME
BECKWITH-WIEDEMANN SYNDROME 
The increased risk for tumor formation in Beck-with-Wiedemann syndrome (BWS) patients is estimated at 7.5% and is further increased to 10% if hemihyperplasia is present. Tumors occurring with the highest frequency include Wilms tumor, hepatoblastoma, neuroblastoma, rhabdomyosarcoma, and ACC.2,199
The genetic basis of BWS is complex. Various 11p15 chromosomal or molecular alterations have been associated with the BWS phenotype and its tumors.200 Abnormalities in this region impact an imprinted domain, indicating that it is more likely that normal gene regulation in this part of chromosome 11p15 occurs in a regional manner and may depend on various interdependent factors or genes. These include the paternally expressed genes IGF2 and KCNQ10T1 and the maternally expressed genes H19, CDKN1C, and KCNQ1.201 Detectable chromosomal abnormalities associated with BWS are extremely rare. 
 MULTIPLE ENDOCRINE NEOPLASIA
MULTIPLE ENDOCRINE NEOPLASIA
The multiple endocrine neoplasia (MEN) disorders comprise at least three different diseases: MEN type 1 (MEN1), MEN type 2A (MEN2A), and MEN type 2B (MEN2B), which are all cancer predisposition syndromes that affect different endocrine organs. 
While MEN1 is caused by mutation in the tumor-suppressor gene, MEN1, MEN2A, and MEN2B are caused by mutations in the protooncogene RET.  The pattern of mutations seen in MEN2 families does not follow the two-hit-hypothesis for tumor suppressor genes: the RET proto-oncogene is not inactivated, and there is no loss of the second allele in the tumors. Thus, the predisposition to cancer in families with MEN2 is based on the inheritance of an activating mutation in the RET proto-oncogene.
The pattern of mutations seen in MEN2 families does not follow the two-hit-hypothesis for tumor suppressor genes: the RET proto-oncogene is not inactivated, and there is no loss of the second allele in the tumors. Thus, the predisposition to cancer in families with MEN2 is based on the inheritance of an activating mutation in the RET proto-oncogene. 
Table 443-2. Hereditary Syndromes Associated with Childhood Neoplasms
MOLECULAR AND CLINICAL SURVEILLANCE FOR CANCER PREDISPOSITION
Continued refinement of molecular analysis for tumor diagnosis, prognosis, and development of novel therapeutic avenues for children with cancer should profoundly influence disease outcomes. The use of molecular screening as a tool for identifying children at risk for the purpose of developing rational clinical surveillance guidelines is more controversial.  Genetic testing should be undertaken only with fully informed consent, including elements of risk assessment, psychological implications of test results (both benefits and risks), risks of employer or insurance discrimination, confidentiality issues and options, and limitations of medical surveillance or prevention strategies. While screening for some mutations, such as RET or RB1, are associated with clearly defined beneficial medical management decisions that lead to improved outcomes, presymptomatic identification of other gene mutations, such as in LFS, are of less obvious clinical benefit. Regardless of the scenario, the complexity of the issues underlies the importance that these patients and families be referred to an experienced multidisciplinary team, including oncologists, geneticists, psychologists, and genetic counselors to facilitate the most appropriate management.
Genetic testing should be undertaken only with fully informed consent, including elements of risk assessment, psychological implications of test results (both benefits and risks), risks of employer or insurance discrimination, confidentiality issues and options, and limitations of medical surveillance or prevention strategies. While screening for some mutations, such as RET or RB1, are associated with clearly defined beneficial medical management decisions that lead to improved outcomes, presymptomatic identification of other gene mutations, such as in LFS, are of less obvious clinical benefit. Regardless of the scenario, the complexity of the issues underlies the importance that these patients and families be referred to an experienced multidisciplinary team, including oncologists, geneticists, psychologists, and genetic counselors to facilitate the most appropriate management.
REFERENCES
See references on DVD.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


