Respiratory Distress Syndrome
EPIDEMIOLOGY
The disease that has defined the history of neonatology is what we now call respiratory distress syndrome (RDS). The enigmatic hyaline membrane disease was uniformly fatal for preterm infants before neonatal care was available.1 The atelectasis, epithelial injury, and accumulation of eosinophilic proteinaceous material in the airspaces were attributed primarily to aspiration or infection. The recognition that the early respiratory failure of some preterm infants could be treated with oxygen in the 1950s contributed to the development of early newborn care centers that have morphed into modern neonatal intensive care units. However, excessive oxygen use resulted in an epidemic of retrolental fibroplasia (now called retinopathy of prematurity).2
The seminal 1959 observation of Avery and Mead3 that the lungs of preterm infants who died of hyaline membrane disease had decreased amounts of surfactant finally identified the primary abnormality that caused the respiratory failure. However, little progress in treatment other than oxygen occurred until the invention of continuous positive airway pressure (CPAP) by Gregory et al4 in 1971 and the concurrent development of mechanical ventilators with positive end-expiratory pressure (PEEP) for infants. Maternal treatment with corticosteroids was described by Liggins and Howie5 in 1972 and to date is the only therapy to prevent RDS. In 1971, Gluck et al6 reported that the L/S (lecithin/sphingomyelin) ratio, a test to measure surfactant components in amniotic fluid, could predict infants at risk of RDS prior to birth. These advances, together with improvements in the general care for preterm infants (better temperature control, fluid therapy, nutrition, physiological monitoring) resulted in high survival rates for the larger and more mature preterm infants, but with the appearance of a new lung disease—bronchopulmonary dysplasia (BPD), caused primarily by oxygen and mechanical ventilation.7 With increased survival, the name of the disease changed from the signature pathologic finding—the hyaline membrane to the physiologic descriptor—respiratory distress.
Experimental studies with premature animals demonstrated that surfactant given via the trachea could improve lung function,8,9 and the first report of successful surfactant treatment of RDS was published in 1980 by Fujiwara et al.10 Surfactants were licensed for general clinical use in 1999–2000 by the Food and Drug Administration (FDA). Subsequent technological improvements in infant ventilators, in the general care of very preterm infants, and in physician skills for the treatment of RDS have resulted in remarkable survival rates with minimal complications for infants with RDS who are greater than 28 weeks’ gestational age at birth.11 Thus, the epidemiology of RDS has changed as the recognition and treatment of the disease has evolved.
In the current era, about 500,000 preterm infants are born in the United States each year, representing about 13% of all live births. Most of these infants are late-preterm infants born between 35 and 37 weeks’ gestational age who have an increased incidence of transient tachypnea of the newborn (TTN) but about a 1% low risk of RDS.12 More immature infants with birth gestations of 32 to 35 weeks have about a 10% RDS incidence.11 The frequency of RDS increases strikingly below gestational ages of 32 weeks to about 50% at 30 weeks’ gestation,13 and the diagnosis of RDS approaches 100% for infants born at less than 28 weeks’ gestation14 (Figure 23-1). The occurrence of RDS is decreased with obstetric interventions to maintain preterm pregnancies at risk of delivery and with antenatal treatments with corticosteroids. As developed in the discussion of differential diagnoses, the epidemiology of RDS does not accurately reflect the true incidence of the disease.
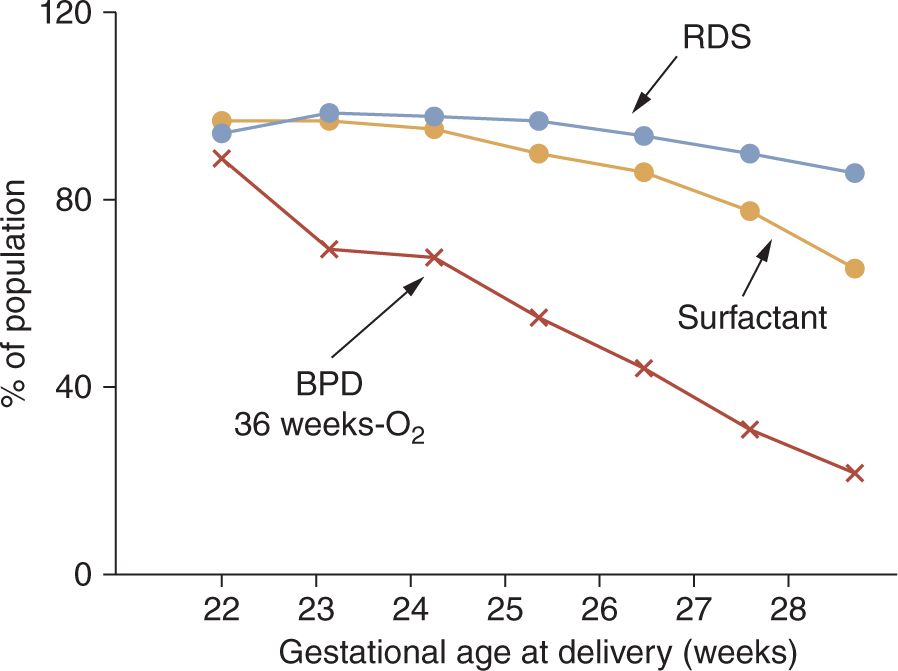
FIGURE 23-1 Respiratory distress syndrome (RDS), surfactant treatments, and bronchopulmonary dysplasia (BPD) relative to gestational age. Data for 9575 infants born at 22–28 weeks’ gestational age from 2003 to 2007 reported by the National Institute of Child Health and Human Development (NICHD) Neonatal Research Network. (Data from Reference 14.)
PATHOPHYSIOLOGY
Surfactant
Respiratory distress syndrome is a disease caused by lung immaturity. The developmental/biochemical problem is surfactant deficiency. The clinical result is respiratory distress caused by inadequate gas exchange from atelectasis, nonuniform aeration of distal lung units that progresses to epithelial injury, proteinaceous pulmonary edema, and ultimately respiratory failure. The central factor is the complex function and metabolism of surfactant in the preterm lung.
Surfactant is a mixture of phospholipids and specific surfactant proteins (SPs) that are produced by type II cells in the alveoli epithelium of air-breathing mammals (Figure 23-2). Surfactant is synthesized in type II cells, packaged into intracellular organelles called lamellar bodies for storage and secretion to the gas exchange surface.15 The composition of surfactant is about 50% phosphatidylcholine with 2 saturated fatty acids, generally palmitic acid, esterified to the glycerol backbone of the molecule.
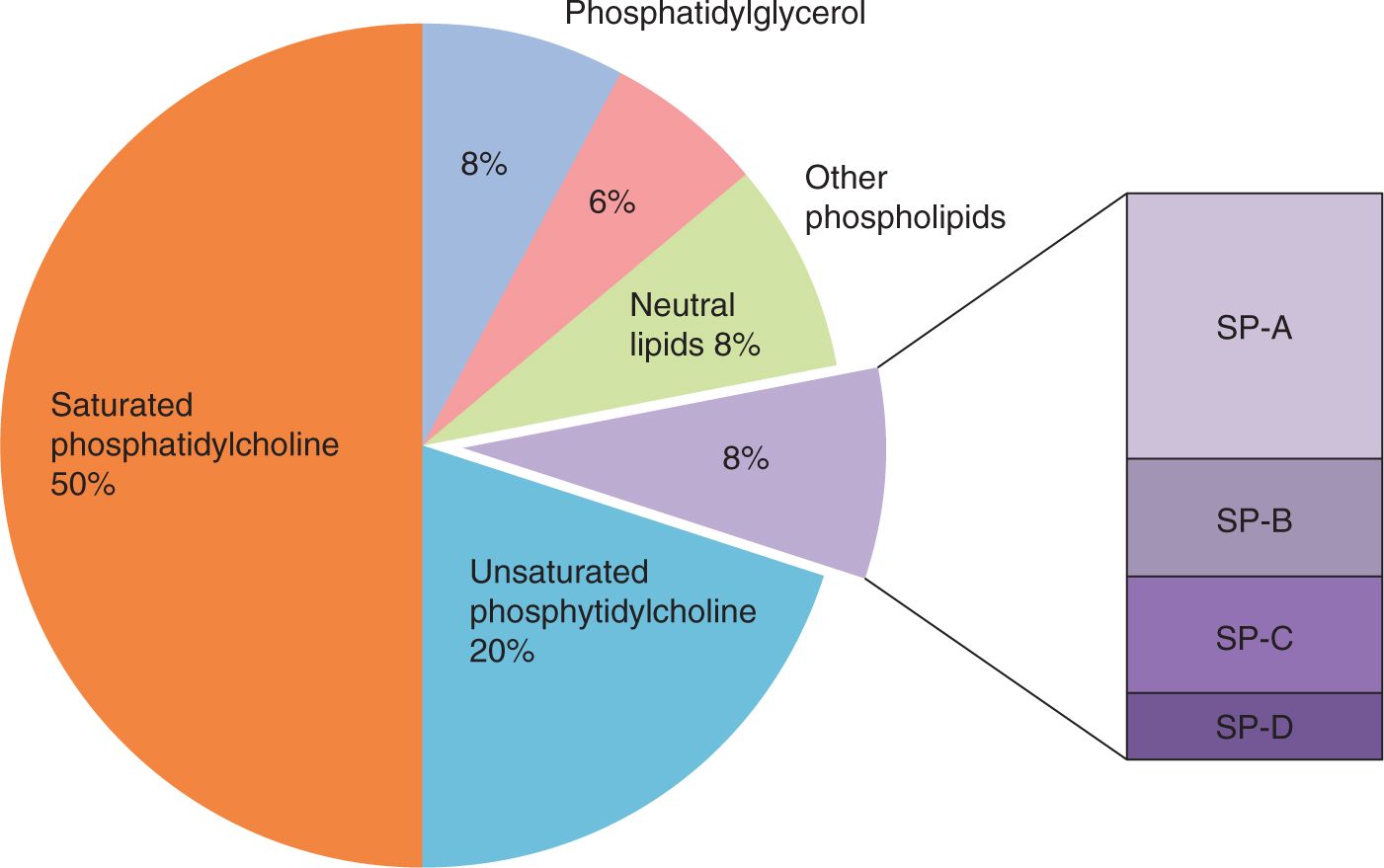
FIGURE 23-2 Composition of surfactant. The major class of surface-active lipids in surfactant are saturated phosphatidylcholines. Surfactant also contains the innate host defense surfactant proteins (SPs) SP-A and SP-D and the lipoproteins SP-B and SP-C.
This “saturated” phosphatidylcholine is extremely surface active as its hydrophilic choline head group solubilizes into water, while the palmitic acids repel water. Surfactant will form a surface film that decreases the equilibrium surface tension of water from about 72 nM/m2 to about 20 nM/m2. With compression of saturated phosphatidylcholine at an air-liquid interface, surface tensions fall to very low values. However, saturated phosphatidylcholine is a solid at 37°C and requires other components to allow it to spread.
Major contributors to the in vivo behavior of surfactant are phosphatidylglycerol, unsaturated phosphatidylcholines, and in surfactant from immature mammals, phosphatidylinositol. These lipids and other minor lipid components interact with 2 small lipophilic SPs, SP-B and SP-C, which are highly conserved across mammalian species.15 The mature form of SP-B is an 8-kDa protein that is required for the intracellular packaging and secretion of the surfactant lipids. The SP-B intercalates in the lipids, facilitating surface adsorption. SP-C is a 4-kDa lipophilic protein that also intercalates with the surfactant lipids to enhance function. A genetic deficiency of SP-C does not cause acute respiratory failure at birth because surfactant lipids with SP-B have good function, although abnormal SP-C can result in interstitial lung disease in later life.16 In contrast, a rare form of lethal RDS primarily in term infants is a genetic deficiency of SP-B because an absence of SP-B disrupts lamellar body formation and the secretion of other surfactant components. The other surfactant-associated proteins, SP-A and SP-D, are collectin family proteins that primarily function as innate host defense proteins. SP-D also regulates surfactant pool sizes, and SP-A makes surfactant more resistant to inactivation by inhibitors (see below).
Early Lung Maturation
Infants have RDS because the synthetic machinery for surfactant is immature, resulting in surfactant deficiency. The lungs of infants are predictably mature when the L/S ratio in amniotic fluid is greater than 2 or when phosphatidylglycerol is detected, which is at about 34–36 weeks for normal pregnancies17 (Figure 23-3). Although measurements of the L/S ratio are primarily of historical interest because easier-to-perform (although less-accurate) tests are in current use, the L/S ratios for normal pregnancies at different gestational ages contrast sharply with the frequency of RDS in clinical practice at these gestational ages. The differences between the timing of normal lung maturation and clinical experience result from the early maturational potential of the fetal lung. As a general perspective, all preterm deliveries should be considered to be abnormal because of the stresses associated with prematurity. There are 2 main pathways to prematurity: (1) vascular developmental abnormalities associated with maternal/fetal diagnoses such as pregnancy-associated hypertension (preeclampsia) and intrauterine growth restriction and (2) intrauterine infection, which can be chronic and clinically silent prior to preterm labor.18 These 2 pathways contribute to most of the very preterm deliveries and contribute to lung maturation earlier in gestation than would occur normally.
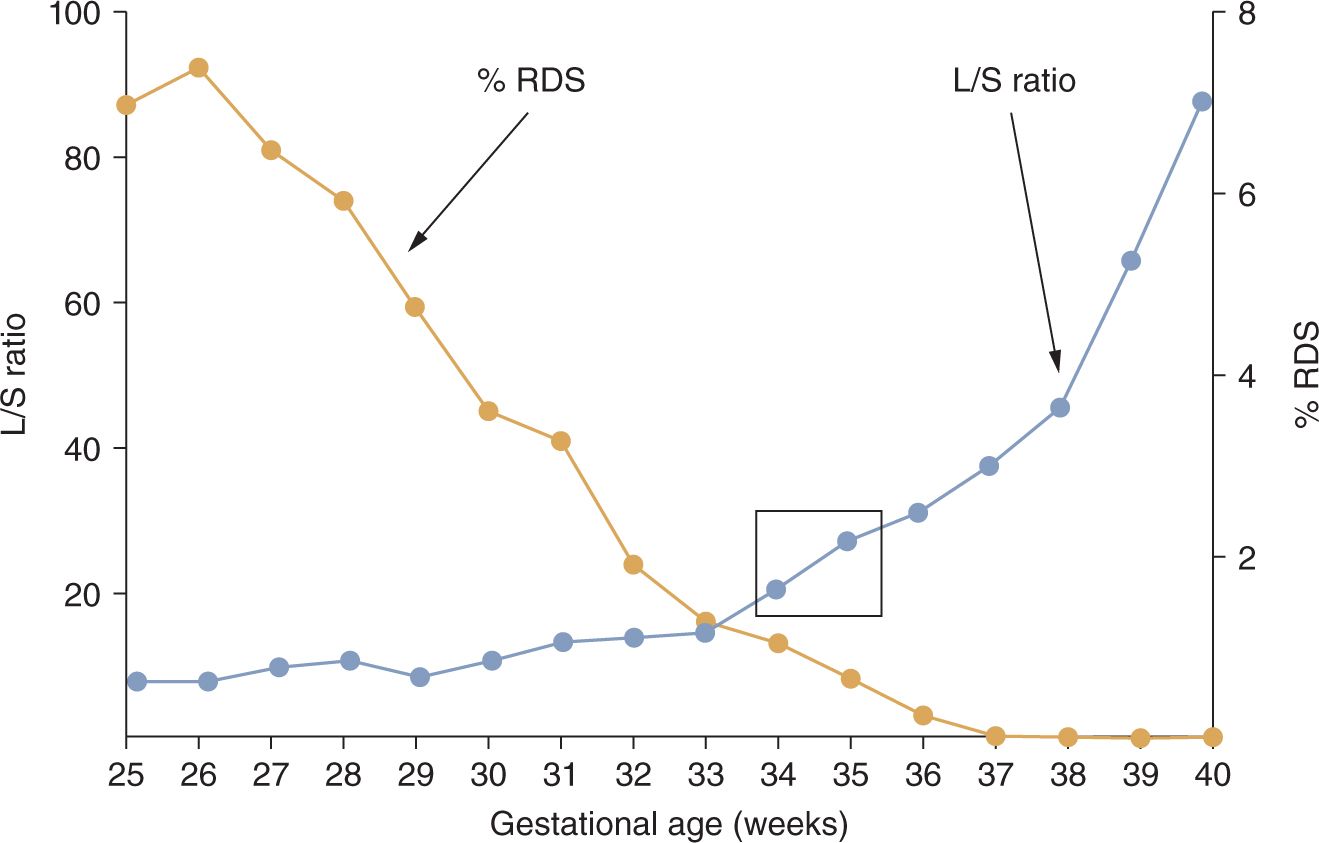
FIGURE 23-3 Incidence of respiratory distress syndrome (RDS) in preterm infants relative to L/S (lecithin/sphingomyelin) ratios for normal pregnancies not delivering preterm. The L/S ratio for normal pregnancies does not indicate maturity with a value greater than 2 until about 35 weeks’ gestational age, as indicated by the box. In contrast, RDS occurs frequently in more preterm infants. (Data from References 6 and 13.)
There are 2 clinically relevant factors that cause early maturation of the fetal lung: corticosteroids and intrauterine exposure to inflammation. Maternal corticosteroid treatments decrease the incidence of RDS for infants born at 28–34 weeks’ gestation by about 50% and are standard of care for women at risk of preterm delivery.19 This simple and inexpensive treatment not only matures the fetal lungs but also has pleotropic and beneficial effects to increase blood pressure, improve postdelivery bilirubin metabolism, decrease intraventricular hemorrhage, and decrease death. The effects on preterm fetal lungs include increased surfactant lipid and surfactant-specific protein synthesis and changes in lung structure to decrease interstitial tissue and increase the potential gas volume.20 Other effects include more rapid clearance of fetal lung fluid after birth and decreased epithelial permeability to protein with ventilation.
The second clinical association with less RDS is fetal exposure to histologic chorioamnionitis. In animal models, fetal exposure to intra-amniotic lipopolysaccharide (a toll-like receptor 4 agonist) or live Ureaplasma parvum (the organism most associated with preterm delivery) results in more induced lung maturation than does maternal corticosteroid treatment (Figure 23-4).21 Curiously, the proinflammation and anti-inflammatory maternal corticosteroids have similar net effects on the fetal lung in that both increase surfactant lipids and proteins and change lung structure to improve gas exchange. In clinical practice, more than 50% of infants born at less than 28 weeks’ gestation have been exposed to histologic chorioamnionitis, and 80% of these pregnancies will be treated with antenatal corticosteroids.22 In both animal models and clinical practice, the 2 exposures decrease RDS and improve outcomes.23 In animal models, the combined fetal exposures to corticosteroids and chorioamnionitis have additive or synergistic effects to increase lung maturation, which should translate clinically to less RDS.24,25 Clinically maternal corticosteroid treatments also decrease RDS in pregnancies with histologic chorioamnionitis.23 Given the available information, the explanations for the frequent birth of infants prior to 34–36 weeks who do not have RDS are fetal stresses that increase fetal cortisol levels, maternal corticosteroid treatments, or fetal exposures to inflammation that mature the fetal lungs.
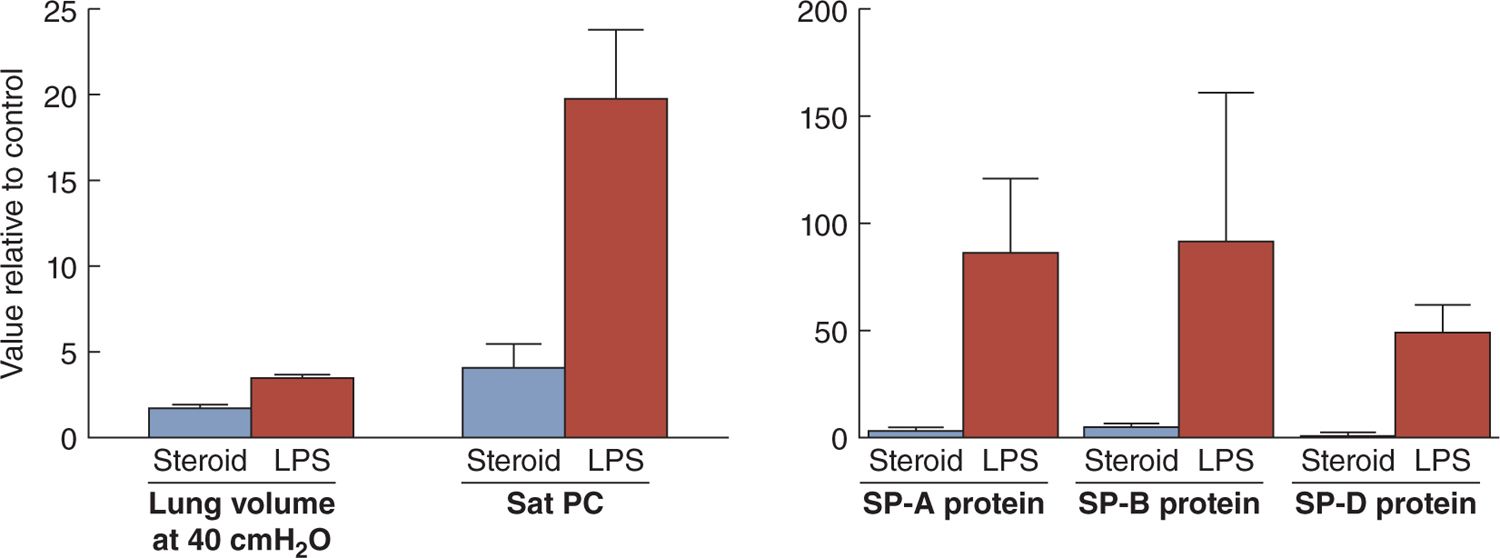
FIGURE 23-4 Increases in lung gas volumes measured at 40 cm H2O pressure and surfactant components measured 7 days after fetal exposure to intra-amniotic lipopolysaccharide (LPS) or betamethasone (steroid) in preterm sheep. The LPS exposure increased lung gas volumes and the surfactant components in surfactant recovered by bronchoalveolar lavage more than did the steroid exposure. All values are expressed relative to a control value of 1. PC, phosphatidylcholine; SP, surfactant protein. (Data from references 20 and 21.)
Surfactant Pool Sizes and Metabolism
As the fetus matures, the distal epithelium of the lung differentiates into immature type II cells and type I cells beginning at about 20 weeks’ gestational age.26 The lamellar body storage granules for surfactant begin to appear after about 22–24 weeks, and an immature surfactant begins to be stored and secreted. This immature surfactant contains less saturated phosphatidylcholine to total phosphatidylcholine, low amounts of the SPs and phosphatidylinositol, and no phosphatidylglycerol. With advancing gestation, more surfactant is stored and secreted and that surfactant is more “mature”—more saturated phosphatidylcholine, more of all of the SPs, and less phosphatidylinositol, and phosphatidylglycerol appears. Surfactant that is secreted is carried into the oral pharynx by the continuously produced fetal lung fluid and fetal breathing. Some of this fluid is swallowed, and some mixes with amniotic fluid, where the increasing amounts of surfactant components can be sampled by amniocentesis for lung maturational testing.
The fetal lung at term contains large amounts of stored surfactant that is secreted just prior to and at delivery in response to redundant stimuli from lung stretch, increases in catecholamines, and purinoreceptor agonists to increase the concentration of surfactant in the fetal lung fluid. This high surfactant concentration facilitates the lowering of surface tensions on menisci of fluid in the airways and facilitates the recruitment of an initial functional residual capacity. The amount of surfactant in the airspaces of term mammals after birth is about 100 mg/kg body weight. In contrast, the adult human lung has about 5 mg/kg surfactant in the airspaces.27 The large amounts in the airspaces at term are presumably a developmental adaptation to ensure the successful transition to air breathing.
Preterm infants destined to develop RDS have greatly decreased stores of surfactant. At birth, the available stores are secreted, and the initial severity of respiratory failure is determined by the pool size of surfactant. Some very preterm infants have almost no surfactant and cannot achieve initial aeration of the lungs without positive pressure recruitment and ventilation. Other, often more mature, infants have a “honeymoon period” during which initial respiratory distress and oxygen needs are minimal, but the RDS progresses over hours. The pool sizes of surfactant estimated by lavages of lungs of infants who died of RDS prior to the era of mechanical ventilation ranged from 0 to 10 mg/kg.28 Preterm lambs or rabbits have minimal RDS if surfactant pool sizes are greater than about 4 mg/kg.29 Measurements in ventilated preterm infants with RDS using stable isotopes supported these values for the amount of surfactant associated with RDS.30
Infants with RDS had progressive respiratory failure in the hours after birth that peaked at about 2–3 days in the presurfactant treatment era. The metabolic behavior of surfactant contributes to this progressive respiratory failure. Over weeks, the fetal lung has accumulated the surfactant pool to be secreted at birth. Once that surfactant is secreted, the accumulation of more surfactant is a slow process that includes new synthesis and recycling of surfactant components from the airspaces back through type II cells for reprocessing and secretion. Measurements of bulk surfactant metabolism in both large animal models and preterm infants with RDS demonstrated that surfactant pool sizes in airspaces take days to increase substantially.
Labeled precursors of saturated phosphatidylcholine appear in the airway surfactant slowly, with a time to maximal labeling (maximal accumulation of newly synthesized surfactant) of about 3 days.31 Once surfactant is in the airspaces, the half-life for clearance is also about 3 days.32 Simultaneously, surfactant components are being recycled efficiently in the preterm lung. The overview is that surfactant components are being slowly, but dynamically, synthesized and recycled because surfactant function depends on newly synthesized surfactant as surfactant is “used” with breathing. Variables known to change surfactant metabolism are lung injury and antenatal corticosteroids, but type of mechanical ventilation (conventional relative to high-frequency oscillation) is a less-important variable.33
Surfactant Inhibition
Another factor contributing to the progression of the symptoms of RDS over several days is the function of the surfactant. The preterm lung at birth that has only surfactant deficiency is immature, but not injured. Fetal lung fluid has very low protein levels, but this fluid must be cleared via ion pumps that are immature. Thus, the surfactant-deficient preterm lung with low surfactant pools has high surface tensions in the small airways. The movement of air-fluid interfaces across epithelial surfaces can disrupt those epithelial barriers, causing the bronchiolar epithelial lesions noted with ventilation of the surfactant-deficient and fluid-filled lung.34 Epithelial disruptions and cell injury increase the permeability of the epithelial barrier to interstitial and intravascular components, resulting in proteinaceous fluid accumulation in the airspace.9
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


