Bronchopulmonary Dysplasia/Chronic Lung Disease
Richard B. Parad
I. DEFINITION.
A National Institutes of Health (NIH) conference proposed definitions for bronchopulmonary dysplasia (BPD), also known as chronic lung disease (CLD) of prematurity, which is a more general term. For infants born at <32 weeks’ gestation who remain in oxygen for the first 28 days at 36 weeks’ postmenstrual age (PMA), mild BPD is defined as no supplemental O2 requirement; moderate BPD is a requirement of supplemental O2 <30%; and severe BPD is a requirement of ≥30% O2 and/or continuous positive airway pressure (CPAP) or ventilator support. For infants born at ≥32 weeks, BPD is defined as a supplemental O2 requirement for the first 28 days with severity level based on O2 requirement at 56 days. A physiologic definition of BPD has been proposed based on SaO2 during a room air challenge performed at 36 weeks (or 56 days for infants >32 weeks) or before hospital discharge, with persistent SaO2 <90%, the cutoff at which supplemental O2 is required. Lung parenchyma usually appears abnormal on chest radiographs. This definition can apply to term babies with meconium aspiration syndrome, pneumonia, and certain cardiac and gastrointestinal (GI) anomalies who require chronic ventilatory support. BPD is associated with the development of chronic respiratory morbidity (CRM).
II. EPIDEMIOLOGY.
Approximately 15,000 new cases of BPD occur in the United States each year. Infants <1,250 g birth weight are most susceptible to developing this condition. Differences in populations (race/ethnicity/socioeconomic status); clinical practices; and definitions account for a wide variation in the rate reported among centers. The relative risk is decreased in African Americans and females. Of infants with birth weight <1,000 g, gestational age <32 weeks, and alive at 36 weeks’ PMA, 44% who require O2 at 36 weeks’ PMA develop CRM (defined as a requirement for pulmonary medications at 18 months corrected age), while 29% without O2 requirement at 36 weeks’ PMA also develop CRM.
III. PATHOGENESIS
Acute lung injury is caused by the combination of O2 toxicity, barotrauma, and volutrauma from mechanical ventilation. Cellular and interstitial injury results in the release of proinflammatory cytokines (interleukin 1β [IL-1β], IL-6, IL-8, tumor necrosis factor-α [TNF-α]) that cause secondary changes in alveolar permeability and recruit inflammatory cells into interstitial and alveolar spaces; further injury from proteases, oxidants, and additional chemokines, and chemoattractants cause ongoing inflammatory cell recruitment and leakage of water and protein.
Airway and vascular tone may be altered. Alveolar development is interrupted, and parenchyma is destroyed, leading to emphysematous changes. Sloughed cells and accumulated secretions not cleared adequately by the damaged mucociliary transport system cause inhomogeneous peripheral airway obstruction that leads to alternating areas of collapse and hyperinflation and proximal airway dilation. Bombesin-like protein, a proinflammatory peptide produced by neuroendocrine cells, is elevated in the urine of infants who subsequently develop BPD. Historically, “old” BPD, as originally reported by Northway in 1967, was described in infants with a mean gestational age of 33 weeks and a birth weight of 2,000 g. Pathology of nonsurvivors showed a predominance of small airway injury, fibrosis, and emphysema. In the postsurfactant therapy era, “new” BPD now predominates, affecting a different population of preterm infants, with a mean gestational age under 28 weeks and birth weight under 1,000 g. For this group, the most significant pathologic finding in nonsurvivors is decreased alveolarization.
In the chronic phase of lung injury, the interstitium may be altered by fibrosis and cellular hyperplasia that results from excessive release of growth factors and cytokines, leading to insufficient repair. Interstitial fluid clearance is disrupted, resulting in pulmonary fluid retention. Airways develop increased muscularization and hyperreactivity. The physiologic effects are decreased lung compliance, increased airway resistance, and impaired gas exchange with resulting ventilationperfusion mismatching and air trapping.
Factors that may contribute to the development of BPD include the following:
Immature lung substrate. The lung is most susceptible before alveolar septation begins. Injury at this stage may lead to an arrest of alveolarization.
Inadequate activity of the antioxidant enzymes superoxide dismutase, catalase, glutathione peroxidase, and/or deficiency of free radical sinks such as vitamin E, glutathione, and ceruloplasmin may predispose the lung to O2 toxicity. Similarly, inadequate antiprotease protection may predispose the lung to injury from the unchecked proteases released by recruited inflammatory cells.
Excessive early intravenous fluid administration, perhaps by contributing to pulmonary edema.
Persistent left-to-right shunt through the patent ductus arteriosus (PDA). Although prophylactic PDA ligation or administration of indomethacin or ibuprofen does not prevent BPD, persistent left-to-right shunt and late PDA closure appear associated with increased BPD risk. However, surgical PDA closure is also associated with increased BPD risk.
Intrauterine or perinatal infection, with cytokine release, may contribute to the etiology of BPD or may modify its course. Ureaplasma urealyticum has been associated with BPD in premature infants, although it remains unclear whether this relationship is causal. Intrauterine Chlamydia trachomatis and other viral infections have been similarly implicated.
Familial airway hyperreactivity is found more commonly in the setting of preterm labor, which confounds the increased risk estimate of both premature and BPD affected infants.
Increased inositol clearance may lead to diminished plasma inositol levels and decreased surfactant synthesis or impaired surfactant metabolism.
An increase in vasopressin and a decrease in atrial natriuretic peptide release may alter pulmonary and systemic fluid balance in the setting of obstructive lung disease.
IV. CLINICAL PRESENTATION
Physical examination typically reveals tachypnea, retractions, and rales on auscultation.
Arterial blood gas (ABG) analysis shows hypoxemia and hypercarbia with eventual metabolic compensation for the respiratory acidosis.
The chest radiograph appearance changes as the disease progresses. In early descriptions of BPD, stage I had the same appearance as respiratory distress syndrome (RDS); stage II showed diffuse haziness with increased density and normal-to-low lung volumes; stage III demonstrated streaky densities with bubbly lucencies and early hyperinflation; and stage IV showed hyperinflation with larger hyperlucent areas interspersed with thicker, streaky densities. Not all infants progressed to stage IV, and some transitioned directly from stage I to stage III. Radiographic abnormalities often persisted into childhood. New BPD is often associated with stage II changes that may evolve if the condition progresses.
Cardiac evaluation. Nonpulmonary causes of respiratory failure should be excluded. Electrocardiogram (ECG) can show persistent or progressive right ventricular hypertrophy if cor pulmonale develops. Left ventricular hypertrophy may develop with systemic hypertension. Two-dimensional echocardiography may be useful in excluding left-to-right shunts (see Chap. 41) and pulmonary hypertension (see Chap. 36). Biventricular failure is unusual when good oxygenation is maintained, and the development of pulmonary hypertension is avoided.
Infant pulmonary function testing (iPFT). Increased respiratory system resistance (Rrs) and decreased dynamic compliance (Crs) have been the hallmarks of BPD. In the first year of life, iPFTs reveal decreased forced expiratory flow rate, increased functional residual capacity (FRC), increased residual volume (RV), and increased RV/total lung capacity ratio and bronchodilator responsiveness, with an overall pattern of mild-to-moderate airflow obstruction, air trapping, and increased airway reactivity.
Pathologic changes are detectable in severe cases by the first few days after birth. By the end of the first week, necrotizing bronchiolitis, obstruction of small airway lumens by debris and edema, and areas of peribronchial and interstitial fibrosis are present. Emphysematous changes and significant impairment in alveolar development result in diminished surface area for gas exchange. Changes in both large airways (glandular hyperplasia) and small airways (smooth muscle hyperplasia) likely form the histologic basis for reactive airway disease. Pulmonary vascular changes associated with pulmonary hypertension may be seen. Arrest of alveolarization is more significant at lower gestational ages.
V. INPATIENT TREATMENT.
The goals of treatment during the neonatal intensive care unit (NICU) course are to minimize further lung injury (barotrauma and volutrauma, O2 toxicity, inflammation), maximize nutrition, and diminish O2 consumption.
Mechanical ventilation
Acute phase. Ventilator adjustments are made to minimize airway pressures and tidal volumes (generally 3—5 ml/kg/breath) while providing adequate gas exchange (see Chap. 30). It is possible that use of patient-controlled ventilator techniques such as patient-triggered breaths, pressure-supported spontaneous
breaths, and volume-targeted patient-triggered breaths may lower BPD risk, although recent trials have not clearly demonstrated this advantage, and some identified increased rates of pneumothorax and mortality. Early use of nasal intermittent positive pressure ventilation (NIPPV) may be more effective than standard nasal CPAP in avoiding need for intubation and surfactant therapy, and NIPPV may also decrease extubation failure rate, although appropriately powered trials are needed.
In most circumstances, we avoid hyperventilation (keeping arterial carbon dioxide tension [PaCO2] at >55 mm Hg, with pH >7.25) and maintain oxygen saturation (SaO2) at 90% to 95% or lower and arterial oxygen tension (PaO2) 60 to 80 mm Hg. We do not routinely use high-frequency oscillatory ventilation because most available evidence suggests that this technique does not prevent BPD in high-risk infants. Early CPAP, with avoidance of mechanical ventilation, and earlier transition from mechanical ventilation to CPAP are management strategies that may be associated with decreased BPD risk.
Chronic phase. Once baseline ventilator settings are established with an PaCO2 not higher than 65 mm Hg, we maintain the ventilator rate without weaning until a pattern of steady weight gain is established.
Supplemental oxygen is supplied to maintain the PaO2 >55 mm Hg. The SaO2 should be correlated with PaO2 in each infant. Several studies published prior to 2007 that evaluated the impact of limiting O2 exposure on retinopathy of prematurity (ROP) risk noted that BPD risk was lower in the groups with lower oximeter saturation ranges. Based on these findings and on other cohort studies, we set oximeter alarm limits at 85% to 93% for infants <32 weeks’ gestation, and then relax the range to 87% to 97% at 32 weeks’ PMA and older. Of caution, the SUPPORT trial (2010) of low (85%-89%) versus high (91%-95%) SaO2 in infants <28 weeks’ gestation revealed a higher mortality rate and no reduction in BPD rate in the low SaO2 group, although severe ROP was less frequent in survivors.
When <30% O2 concentration is required by hood, we supply O2 by nasal cannula. If adequate SaO2 cannot be maintained on <1 L/min of flow, we restore hood O2. We use a flowmeter that is accurate at low rates, and gradually decrease the flow of 100% O2 while maintaining the appropriate SaO2. Alternatively, flow can be decreased to the lowest marking on the flowmeter, as tolerated, and then O2 concentration can be decreased. Estimates of the actual concentration of O2 delivered to the lungs by nasal cannula at different flows of 100% O2 have been generated by hypopharyngeal measurements (see Fig. 34.1). SaO2 should remain >90% during sleep, feedings, and active periods before supplemental O2 is discontinued.
Surfactant replacement therapy decreases the combined outcome of CLD or death at 28 days of age, although it has made little or no impact on the overall incidence of CLD. Meta-analyses suggest that the incidence is decreased in larger premature infants but is higher in smaller premature infants who would have died without surfactant therapy (see Chap. 33). Late surfactant exhaustion may contribute to the development of BPD; an ongoing trial is testing late surfactant dosing in infants with decompensation or persistent requirement for mechanical ventilation.
PDA. We consider treatment of a hemodynamically significant PDA in infants who have respiratory decompensation or cannot be weaned from mechanical ventilation (see Chap. 41).
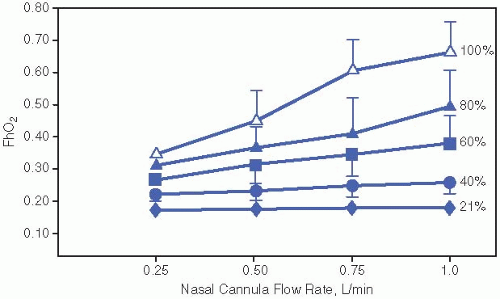
Figure 34.1. Approximate conversion from nasal cannula flow FiO2 to hypopharyngeal FiO2 (FhO2). FiO2, fraction of inspired O2Get Clinical Tree app for offline access
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


