Bleeding Disorders
Gregory E. Plautz
The evaluation of a child or an adolescent for a bleeding disorder rests on an assessment of whether the bleeding or bruising is abnormal or is within the range of normal. It is typical for toddlers to have small bruises on extensor surfaces and for children to have several episodes of epistaxis. Likewise, menarche frequently raises concerns about irregular bleeding of variable amounts that can lead both parents and patients to evaluation by the pediatrician. As with all evaluations, history, family history, physical examination, and the judicious use of a limited number of key laboratory tests are usually sufficient to diagnose most pediatric bleeding disorders.
This chapter discusses the common pediatric abnormalities in the three components of the coagulation system:
Platelet disorders
Coagulopathies
Thrombophilia
Discussion of platelet abnormalities includes both quantitative defects and functional defects. Similar to many pediatric diseases, there are congenital defects that usually present early in infancy and acquired defects that can present at any age. Most patients harboring abnormalities in plasma components have either von Willebrand disease (vWD) or hemophilia. Finally, inherited disorders that cause thrombophilia, an abnormal propensity to blood clot formation, are briefly described.
NORMAL COAGULATION CASCADE AND LABORATORY SCREENING TESTS
It is helpful to overview briefly the process of coagulation and its relation to the most commonly ordered coagulation tests (Fig. 57.1). The coagulation factors are enzymes that normally exist in an inactive form in plasma. Upon enzymatic cleavage of a portion of the molecule, there is a change in its conformation that activates its proteolytic function. The activated coagulation factors then interact with the next downstream substrate, thereby forming a cascade that amplifies the response and ultimately generates a fibrin plug. It is important to realize that coagulation normally occurs on a negatively charged phospholipid surface provided by the platelet or endothelial plasma membrane.
When a blood vessel is injured, the endothelial surface is disrupted, causing two arms of the coagulation system to become activated. First, platelets are exposed to subendothelial collagen, activating surface glycoproteins and von Willebrand factor (vWF). Second, tissue factor (TF) in the subendothelial space comes in contact with the small amounts of plasma factor VII that exist in an activated form (designated VIIa). The activated platelets clump to form a physical obstruction. In addition, platelets degranulate, releasing substances that cause vasoconstriction, and thereby staunching the blood flow. Exposure of factor VIIa to TF on a phospholipid surface causes cleavage and activation of factor X (designated Xa). Factor Xa, factor VIIa, calcium, and phospholipids combine to form the prothrombinase complex. The most important step is the conversion of prothrombin (factor II) to thrombin. Thrombin acts on fibrinogen, which is a relatively abundant substrate in plasma, to generate fibrin monomers. These fibrin monomers are subsequently cross-linked by factor XIII to form a stable clot.
The prothrombin time (PT) is performed by adding recombinant TF, a phospholipid complex, and calcium to anticoagulated (citrated) plasma. TF reacts with the small amount of factor VIIa to initiate the “extrinsic” pathway. It is called “extrinsic” because it relies on TF, which is normally extrinsic to the vasculature. The PT is dependent on factors VII, X, V, II (prothrombin), and fibrinogen. Several of these factors (II, VII, and X) are vitamin K dependent, and the PT is often used to monitor warfarin therapy. The International Normalized Ratio (INR) is an adjusted PT that was developed to compensate for variability in different batches of reagents to warfarin inhibition, and it represents patient PT/control PT. The INR shows much less interlaboratory variability for patients on warfarin therapy.
The activated partial thromboplastin time (aPTT) is performed by adding phospholipids, calcium, and a particulate substance to citrated plasma. This serves to activate the
“intrinsic” pathway because all of the factors are intrinsic to plasma. The earliest step in the cascade involves interaction between the phospholipid surface and “contact” factors, including the following: factor XII, high-molecular-weight kininogen (HMWK), prekallikrein, and factor XI. The initiating factors then cleave to factor IX, thereby activating it (factor IXa). The phospholipid surface also serves as a platform on which factors IXa and VIII combine to activate factor X. Activated factor X (Xa) then combines with factor Va to generate the prothrombinase complex as described in the preceding text.
“intrinsic” pathway because all of the factors are intrinsic to plasma. The earliest step in the cascade involves interaction between the phospholipid surface and “contact” factors, including the following: factor XII, high-molecular-weight kininogen (HMWK), prekallikrein, and factor XI. The initiating factors then cleave to factor IX, thereby activating it (factor IXa). The phospholipid surface also serves as a platform on which factors IXa and VIII combine to activate factor X. Activated factor X (Xa) then combines with factor Va to generate the prothrombinase complex as described in the preceding text.
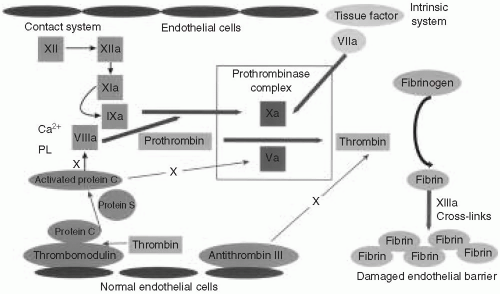 Figure 57.1 Components of the coagulation system. PL, phospholipid. |
Feedback Inhibition of the Coagulation Cascade
It is physiologically important to restrict clot formation to the immediate vicinity of the endothelial damage. Consequently, there is feedback through an elaborate antithrombotic system to balance the coagulation cascade. When thrombin is generated, it binds to thrombomodulin and together with protein S enzymatically activates protein C. Activated protein C inactivates factors VIIIa and Va. There is a second system that is maintained in a highly activated state. It consists of the protein antithrombin III, which is bound to heparin sulfate on the surface of intact endothelium. When factors Xa and thrombin drift into a zone of intact endothelium, they are degraded rapidly by antithrombin III. This prevents clot propagation beyond the area of damaged endothelium.
PLATELET DISORDERS
The first step in the evaluation of thrombocytopenia involves determination of whether the platelet count is abnormal. There is a wide range of normal, from 150,000 to 450,000 mm3, but no age-dependent variation in normal level. The platelet count determines the level of severity: A platelet count >50,000/mm3 is mild thrombocytopenia with minimal bleeding risk in the absence of major trauma or surgery. A platelet count 20,000 to 50,000/mm3 is moderately severe with risk of bleeding from contact sports. A platelet count of <20,000/mm3 is considered severe thrombocytopenia and carries an increased risk of spontaneous bleeding. An enlarged spleen can sequester platelets, but it normally does not cause the platelet count to drop <50,000/mm3. The next step is to determine whether there is decreased production or increased destruction of platelets. The normal platelet survival time is approximately 7 days. Similar to red blood cells in anemia, recently produced platelets have an increased volume (mean platelet volume [MPV]) and have residual mRNA. Some laboratories can provide a “reticulated platelet” count, which if low implies a production defect, and if high implies destruction.
Immune Thrombocytopenic Purpura
Immune thrombocytopenic purpura (ITP) is caused by autoantibodies binding to platelets with subsequent clearance by the spleen. In children, the peak incidence is from 2 to 5 years of age. The clinical onset is acute, but often there is a subclinical viral prodrome 1 to 3 weeks prior to the onset of clinical findings. The history is notable for a lack of previous bleeding or bruising tendency. Physical signs include bruising in locations other than extensor surfaces and petechiae (1- to 3-mm nonblanching nontender bright red-purple skin lesions). Occasionally, mucous membrane petechiae (wet purpura) are present. Hepatosplenomegaly and/or diffuse lymphadenopathy are not associated with ITP and
if present should prompt a workup for leukemia. Microscopic hematuria is frequently present. The laboratory workup should include:
if present should prompt a workup for leukemia. Microscopic hematuria is frequently present. The laboratory workup should include:
Complete blood count with differential
Reticulated platelet count (if available)
Type and screen
Coombs test
Examination of a peripheral blood smear to exclude microangiopathic destruction, to rule out hemolytic uremic syndrome and thrombotic thrombocytopenic purpura
A bone marrow aspiration is not routinely performed unless particular physical or laboratory findings are suspicious for leukemia or aplastic anemia.
Clinical Course and Management of Immune Thrombocytopenic Purpura
ITP is usually a self-limited disease with 90% of cases resolving within 6 months. The goal of therapy is to prevent life-threatening hemorrhage, which occurs in 0.5% to 2% of cases. Platelet transfusions are not routinely performed because the antibodies are usually directed against common antigens. However, platelet transfusions and emergency splenectomy have been performed for life-threatening bleeding. Patients with platelet count <10,000/mm3 should be hospitalized.
Patients with ITP who are Rh-positive may be treated with anti-Rh globulin, (WinRho 50-75 μg/kg intravenously for one dose). It may be necessary to repeat the dose in 3 to 4 weeks. The anti-Rh globulin binds to red blood cells and overwhelms binding sites in the reticuloendothelial system, thereby decreasing the removal of platelets. Anticipate a drop in the hemoglobin concentrations by 1 to 2 g/dL. Anti-Rh should be used with caution if the baseline hemoglobin concentration is <8 g/dL.
Patients who are Rh negative may be treated with intravenous immune globulin; the recommended dose is 0.8 g/kg. Again, this dose may need to be repeated depending on the response.
Alternative therapies for ITP include a high-dose of methylprednisolone 30 mg/kg per day for 3 days or prednisone 2 mg/kg per day for 2 weeks with subsequent slow taper.
Chronic Immune Thrombocytopenic Purpura
Chronic ITP is defined as ITP lasting for >6 months. There is a 3:1 female to male preponderance, and the typical patient is an adolescent girl. Chronic ITP is often associated with a positive antinuclear antibody and may be the initial manifestation of systemic lupus erythematosus or other connective tissue disorder. Splenectomy is effective in 75% of cases. Rituximab (anti-CD20 mAb) can be used for severe chronic ITP but can cause immunosuppression because of depletion of B lymphocytes.
Neonatal Thrombocytopenia
Thrombocytopenia is common among “ill” neonates and preterm infants and may occur secondary to birth asphyxia, meconium aspiration, respiratory distress, necrotizing enterocolitis, or infection.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


