Dysmorphic Syndromes
George E. Tiller
Dysmorphology is the art and science of abnormal physical development. It is often subjective, and distinguishing between familial traits and true dysmorphic features is sometimes difficult. A recognizable pattern of human malformation is dubbed a syndrome. Some syndromes are associated with a single underlying genetic defect or chromosomal anomaly, others have a multifactorial basis (genetic plus environmental), and still others have no apparent genetic basis at all, being simply birth defects. Genetic disorders contribute significantly to morbidity and mortality at all ages. More than 3500 mendelian (single-gene) disorders have been catalogued. It has been estimated that 1% to 2% of newborns have significant birth defects. Although individually rare, genetically related disorders are responsible for one third of all pediatric hospital admissions. We must also appreciate that many genetic disorders are insidious in onset and may present to the clinician in a wide range of age groups.
A three-generation family history is an essential component of the evaluation of a possible genetic disorder. Questions about ethnicity and possible consanguinity are a routine part of this process. The family history is followed by a review of the patient’s medical history and then by a careful, objective physical examination. Anthropometric graphs from appropriate texts can remove some of the subjectiveness involved in determining the disproportionality of body parts. Once a differential diagnosis has been constructed, specific testing can begin, if available. Genetic counseling is the last step in the clinical evaluation, once all the information available has been analyzed.
The objectives of this chapter are to help the reader:
Distinguish different mechanisms underlying congenital anomalies
Describe the clinical features and molecular basis (if known) of the more common dysmorphic syndromes
CATEGORIES OF CONGENITAL ANOMALIES
Malformation: A morphologic defect resulting from an intrinsically abnormal developmental process. Causes may be chromosomal, genetic (a single gene), teratogenic, or idiopathic. Examples are cleft lip, cleft palate, congenital heart defects, and neural tube defects.
Deformation: An abnormal form, shape, or position of a body part caused by mechanical forces. The cause is often uterine constraint. Examples are craniosynostosis and clubfoot.
Disruption: Destruction of a previously normally forming body part. Causes include amniotic bands and vascular disruption. Examples are limb reduction defects and gastroschisis.
Dysplasia: Abnormal organization or proliferation of cells into a tissue. Causes include some single-gene defects. Examples include ectodermal dysplasias and skeletal dysplasias.
Sequence: A cascade effect resulting from a single embryologic event that may affect surrounding tissues. Examples include Pierre-Robin sequence, amnion rupture, oligohydramnios, and DiGeorge syndrome.
WHEN TO CONSIDER A DYSMORPHIC SYNDROME
Dysmorphic syndromes must be considered in the presence of:
More than one anomaly present
Phenotypic variation from parents and siblings
Abnormal growth: prenatal (and sometimes postnatal) growth retardation (or overgrowth)
Neurologic symptoms (e.g., seizures, mental retardation [MR], abnormal muscle tone, deafness, blindness, speech delay with or without motor delay)
EXAMPLES OF MORE “COMMON” GENETIC SYNDROMES
Achondroplasia
Frequency: Most common nonlethal skeletal dysplasia (1 in 26,000)
Inheritance: Autosomal dominant (80% new mutations)
Features: Short-limbed dwarfism, macrocephaly with frontal bossing and saddle nose, infantile gibbus progressing to lumbar lordosis, trident hand, and tibial bowing (Fig. 42.1)
Complications: Infantile hypotonia, apnea secondary to tight foramen magnum, ventriculomegaly, and cervical and lumbar spinal cord compression
Primary defect: Fibroblast growth factor receptor 3
Marfan Syndrome
Frequency: 1 in 10,000
Inheritance: Autosomal dominant (>15% new mutations)
Features: Tall, lanky habitus; dolichocephaly and high, arched palate; arachnodactyly, joint laxity; pectus deformity, scoliosis; myopia, ectopia lentis; mitral valve prolapse, aortic root dilatation; dural ectasias (Figs. 42.2, 42.3 and 42.4)
Complications: Joint instability, visual impairment, aortic insufficiency, aneurysmal rupture causing sudden death, spontaneous pneumothorax, and pulmonary blebs
Treatment: Atenolol or other cardioselective β-blocker; losartan
Primary defect: Fibrillin, a connective tissue protein
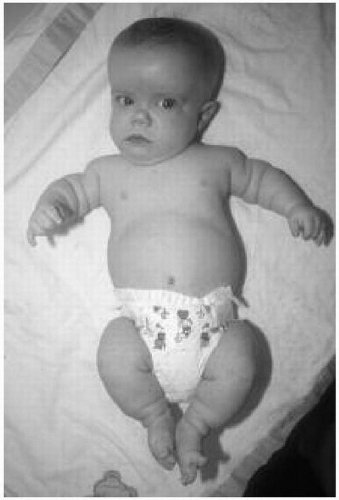 Figure 42.1 Six-month-old boy with achondroplasia. Note the macrocephaly, frontal bossing, upturned nares, and short limbs. |
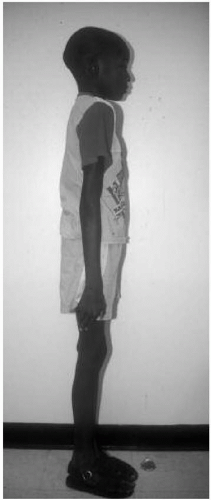 Figure 42.2 Nine-year-old boy with Marfan syndrome. Note the tall, thin stature. |
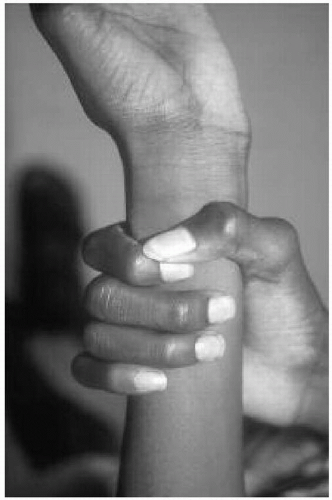 Figure 42.3 Nine-year-old boy with Marfan syndrome. Note the positive wrist sign. |
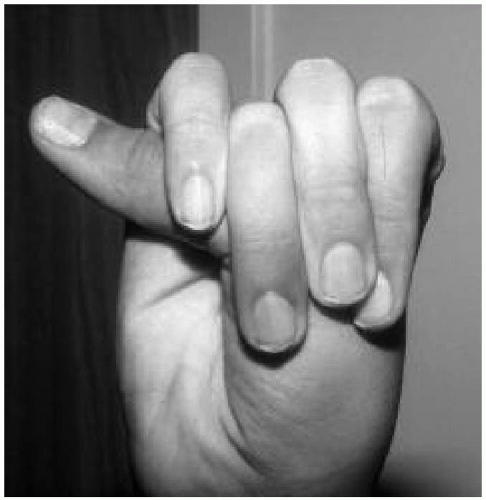 Figure 42.4 Twelve-year-old boy with Marfan syndrome. Note the positive thumb sign. |
Ehlers-Danlos Syndrome
Frequency: 1 in 10,000
Inheritance: Autosomal dominant (mostly), rarely autosomal recessive
Features: Skin hyperelasticity (Fig. 42.5), poor wound healing, excessive bruising; joint laxity (Fig. 42.6), pectus deformity, and scoliosis
Complications: Joint instability; aneurysmal rupture causing sudden death (type IV or arterial form only)
Primary defects: Type III or V collagen; lysyl hydroxylase; others unknown
Remarks: Ten known clinical types of Ehlers-Danlos syndrome, caused by defects in different genes and with varying severity
Osteogenesis Imperfecta
Frequency: 1 in 20,000
Inheritance: Autosomal dominant, rarely autosomal recessive
Features: Vary from mild to lethal (perinatal); osteoporosis, bone fragility causing pathologic fractures (Fig. 42.7); dentinogenesis imperfecta (Fig. 42.8); Wormian bones in occiput; blue sclerae; hearing loss
Complications: Pulmonary hypoplasia, death (type II); scoliosis, progressive deformities (type III); short stature (type IV)
Primary defect: Type I collagen
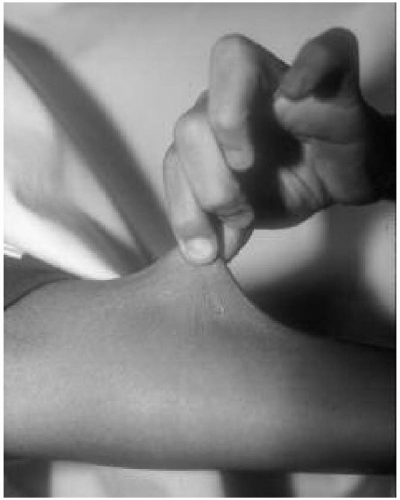 Figure 42.5 Eight-year-old boy with Ehlers-Danlos syndrome (type VIII). Note the skin hyperelasticity on the forearm. |
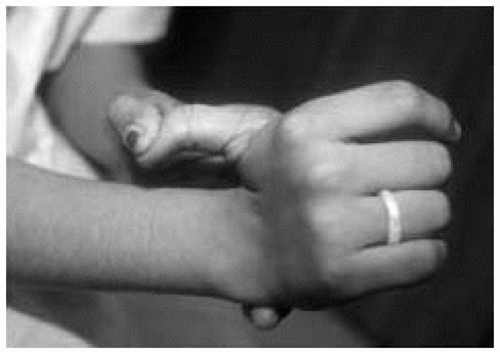 Figure 42.6 Eighteen-year-old woman with Ehlers-Danlos syndrome (type II). Note the joint hyperextensibility. |
Anhydrotic Ectodermal Dysplasia
Frequency: 1 in 10,000
Inheritance: X-linked; female carriers mildly affected
Features: Hypotrichosis, hypodontia, anhydrosis, heat intolerance, atopy, short stature, and mild MR
Primary defect: Ectodysplasin, a transmembrane protein
Remarks: Numerous other ectodermal dysplasia disorders are known!
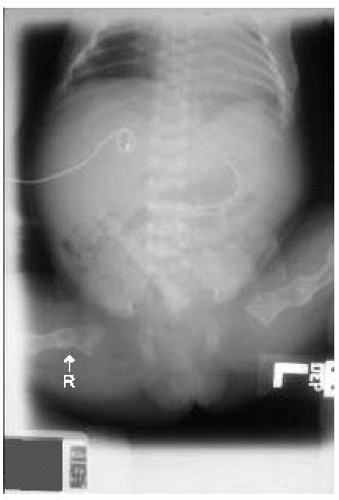 Figure 42.7 Radiograph of term newborn girl with osteogenesis imperfecta. Note the diffuse osteopenia, healed femoral fracture (L), and displaced acute femoral fracture (R). |
 Figure 42.8 Five-year-old girl with osteogenesis imperfecta. Note the dentinogenesis imperfecta. |
Holt-Oram Syndrome (Heart-Hand Syndrome)
Frequency: 1 in 100,000
Inheritance: Autosomal dominant, variable expression
Features: Congenital heart disease (atrial septal defect > ventricular septal defect); finger-like or absent thumb (Fig. 42.9); radial hypoplasia
Primary defect: TBX5 gene, a transcription factor
Thanatophoric Dysplasia
Frequency: 1 in 50,000 (most common lethal skeletal dysplasia)
Inheritance: Sporadic, dominant-acting mutation
Features: Short-limbed dwarfism, curved long bones, narrow thorax, and flattened vertebrae (platyspondyly)
Complications: Death resulting from pulmonary hypoplasia
Primary defect: Fibroblast growth factor receptor 3 (allelic with achondroplasia)
Treacher-Collins Syndrome (Mandibulofacial Dysostosis)
Frequency: 1 in 10,000
Inheritance: Autosomal dominant
Features: Malar and mandibular hypoplasia, external and middle ear anomalies, downward-slanting palpebral fissures (Figs. 42.10 and 42.11), eyelid colobomata, and cleft palate
Complications: Feeding difficulties, conductive hearing loss, MR (5%), and congenital heart disease (10%)
Primary defect: TCOF1 (aka treacle), a nucleolar trafficking protein
Remarks: Several features in common with Goldenhar syndrome, but anomalies in Treacher-Collins (TC) syndrome are usually symmetric, and TC syndrome is dominantly inherited
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


