Biliary Tract Anomalies
BILIARY ATRESIA
Epidemiology
Although biliary atresia is a rare disorder, it is the most common surgically correctable liver disorder in infancy. The most accurate estimates of national prevalence come from the United Kingdom and France, where 1 in 17,000–19,000 live-born infants are affected.1,2 East Asian countries are most commonly affected, with a reported frequency of 1 in 5000 in Taiwan.3Other reported estimates are 1 in 15,000 in the southeastern United Sates4 and 1 in 19,000 in the Netherlands.5 Within these regions, there appears to be a higher incidence of the disease in non-white populations (African American, French Polynesian, and Chinese) and among females (1.25:1).6,7
Up to 20% of all cases of biliary atresia are associated with other anatomical abnormalities, suggesting that the pathologic process begins in the embryonic period. The most common congenital cluster of malformations is biliary atresia splenic malformation (BASM) syndrome, seen in approximately 10% of European and US series. BASM includes biliary atresia in association with polysplenia (90%), situs inversus (50%), and unusual vascular anomalies. In utero, there may also be an association with maternal diabetes and genetic anomalies such as trisomy 18 and 21.
In the other 80% of neonates with biliary atresia, the disorder appears to be a sporadic event with an unknown etiology, and the pathological obliterative process begins later in the perinatal period. Classic genetic inheritance is not supported by any clinical evidence as the disorder is rarely seen within families and, when seen in twins, is rarely concordant.8 Some studies have suggested time-space clustering of cases and seasonal variation, with the majority of cases occurring in the fall and winter months (December–March),9,10 but large population-based studies from France, Sweden, and Japan have failed to identify significant seasonal or time-space clustering.9–12 After controlling for geographic and racial factors, associations have been made with advanced maternal age and increased parity, but not with smoking, maternal age, education, alcohol use, folic acid intake, gravidity, parental income, infant sex, preterm birth, infant birthweight, or plurality.5,9,12
Pathophysiology
Although histopathologic features of biliary atresia have been extensively studied in surgical specimens from excised extrahepatic biliary systems of infants, the pathogenesis of this disorder remains an unanswered question. Because some infants with biliary atresia are born with other congenital malformations, early studies postulated the etiology was a congenital malformation of the biliary ductal system. However, the majority of patients have isolated biliary atresia with progressive inflammatory lesions on histology occurring in the perinatal period. Therefore, it might be that biliary atresia represents a final common phenotypic pathway of neonatal liver injury caused by a diverse group of etiologies, including viral, inflammatory, immune dysregulation, or toxic exposure in genetically predisposed individuals5 (Figure 41-1).
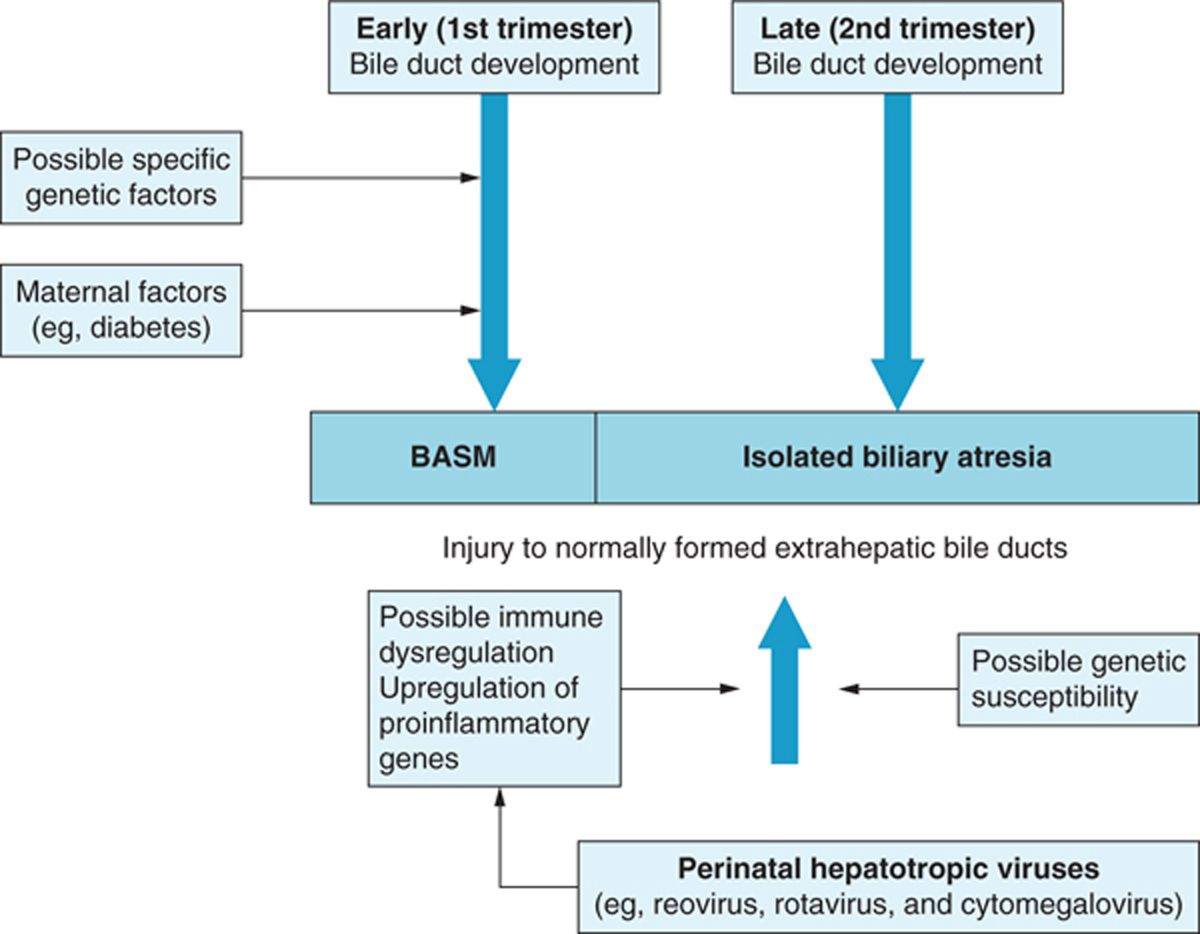
FIGURE 41-1 Possible causal relations in biliary atresia. BASM, biliary atresia splenic malformation. (Reprinted from The Lancet, Hartley, Davenport, and Kelly.5)
Genetics
There is a growing body of evidence supporting the involvement of specific genes in both embryonic (somatic mutations of genes involved in morphogenesis of the biliary tree) and perinatal (genetic predisposition to an aberrant immune response to exogenous stimuli) biliary atresia. In the embryonic form, underlying genetic mutations may lead to morphologic abnormalities. In mice, a mutation of the INVS gene can cause morphologic change resembling biliary atresia.13 In these mice, morphological analysis of the hepatobiliary system by Trypan blue cholangiography and technetium 99m-labeled tracer demonstrated a defect in patency of the extrahepatic ductular system and absent biliary excretion.14 Inconsistent with the diagnosis, however, is the complete absence of inflammation or necrosis within hepatic parenchyma and absence of inflammation and fibrosis of the extrahepatic biliary tree.14,15 In addition, human studies of the INVS gene did not support the finding of morphologic defects.16 Other genetic etiologies of morphologic changes in mice have been proposed, including abnormalities in (HNF)-6 and HNF-1B.17,18 and constitutional or delayed inactivation of genes Hes1, Foxf1, and Foxm1b,19–21 but these are still being investigated. In the perinatal form, genetics may be the foundation for further infectious or inflammatory insult. For example, alterations in genes, including CFC1, ICAM1 (intercellular adhesion molecule 1), macrophage migration inhibitory factor gene, CD14 endotoxin receptor gene, and hepcidin antimicrobial peptide gene, can speed the progression of biliary fibrosis.22–25
Viral
The possibility of a viral infection acting as the initiating event for biliary atresia was first suggested by Landing, who saw biliary atresia along a continuum with choledochal cysts and neonatal intrahepatic cholestasis, all of which could be linked by a shared infectious insult.26 Since the introduction of this concept, viral pathogenesis of nonsyndromic causes of biliary atresia has been studied in animals and humans. Inoculation of mice with rotavirus strains RRV and SA11-FM,27 reovirus, and cytomegalovirus (CMV)28,29 has resulted in jaundice with intrahepatic histology similar to biliary atresia. In humans, an interesting study of viral infection in biliary atresia was conducted by Rauschenfels and coworkers,30 who used wedge liver biopsy samples obtained from 74 infants at Kasai portoenterostomy to look for a panel of DNA and RNA hepatotropic viruses and Mx protein, a marker of inflammation secondary to viruses. A third of infants had evidence of viruses, and this finding increased with age, suggesting that the viral component may be a secondary insult to the specific cause of biliary atresia. Ninety percent of infants expressed Mx protein, suggesting that there is ongoing inflammation secondary to the immune response, perhaps mounted from a viral infection.30 Additional supporting evidence of a viral etiology are hepatitis B virus antigens detected in the liver of infants with biliary atresia in Japan,31 prevalence of antibodies against reovirus type 3 and the detection of the virus in hepatobiliary specimens of patients with biliary atresia,32–35 and a study demonstrating that reverse transcription–polymerase chain reaction (RT-PCR) identified reovirus in 55% of hepatobiliary samples from patients with biliary atresia.36 Also suggestive of a viral role is that CMV infection in infants with biliary atresia has a severe clinical course, with low rates of jaundice clearance and raised rates of cholangitis and liver fibrosis.37 However, negating this suggestion of viral etiology is that viral particles in the liver or biliary tract of infected infants have not been reproducible across studies conducted at different time points or in different locations. This inconsistency and lack of reproducibility may be due to the clearance of direct viral traces by an inflammatory reaction or simply lack of consistent viral etiology.38
Inflammation/Immunology
Interestingly, when an infant has biliary atresia, there is a pronounced inflammatory response in both the liver and circulation. In the liver, mononuclear cells infiltrate the periductal space, and the vascular and biliary epithelium have increased expression of HLA-DR and intracellular adhesion molecules such as ICAM1 and E selectin.39 Simultaneously, the circulation has increased numbers of soluble inflammatory adhesion molecules and cytokines, even when a successful Kasai portoenterostomy has been performed. Although it appears that the bile duct damage is most likely lymphocyte-mediated biliary inflammation, the trigger for this response remains unknown.
Given the histologic features of progressive inflammation, various studies have implicated immune dysregulation, either as a primary disorder or as the result of infectious or genetic triggers, as a potential link with biliary atresia. In patients with biliary atresia, inflammatory cytokines such as interlukin 2, interleukin 12, interferon γ (IFN-γ), and tumor necrosis factor α (TNF-α) are upregulated along with Kuppfer cells, natural killer (NK) cells, CD3+ and CD8+ T cells, and CXCR3+ cells.40 Of note, the oligoclonal expansion of CD4+ and CD8+ T cells within the liver and extrahepatic bile ducts is suggestive of a response to specific antigenic stimulation.41 An example of a specific infectious trigger is the development of biliary atresia in mice after inoculation with rotavirus is increased when type I IFN receptor is inactivated.42 and the tissue-specific hepatobiliary inflammation induced by dysregulation of gamma interferon after inoculation with rotavirus.43 Genetic triggers of immune dysregulation may also play a role. Coordinated activation of genes involved with lymphocyte differentiation, particularly those associated with T helper 1 immunity, has been identified in liver samples from infants with biliary atresia.5 Polymorphisms that enhance expression of the CD14 gene, which plays a role in the recognition of bacterial endotoxin, have been associated with biliary atresia.5 Gene expression microarrays of RNA from extrahepatic tissue and gallbladders in rotavirus-induced murine models of biliary atresia have shown upregulation of many genes regulating immunity.44 Microarrays from human samples have also shown an overexpression of immune regulatory genes.45
Graft-vs-host issues may also play a role, as demonstrated by a high concentration of maternal chimeric cells that have been found in the portal and sinusoidal areas of patients with biliary atresia, suggesting that maternal lymphocytes cause bile duct injury through a graft-vs-host immune response.46
Toxic Insult
The only supportive patient-based evidence for the role of a toxic insult as a causative factor of biliary atresia is the time-space clustering of cases. In Australia between 1964 and 1988, there were unusual outbreaks of hepatobiliary injury in lambs and calves in New South Wales. However, despite localized outbreaks, there were no causative phytotoxins or myotoxins discovered through exhaustive investigation.47
Differential Diagnosis
There is a high degree of overlap in clinical, radiologic, and histologic characteristics of biliary atresia and other causes of hepatitis in the neonatal period. The differential is broad and includes structural, genetic, infectious, and metabolic conditions.48 Some of these conditions include anatomic (choledochal cyst, bile duct stenosis, sclerosing cholangitis of the newborn, cholelithiasis, tumors/masses); infectious (viral, bacterial); metabolic/genetic (Alagille syndrome [AGS], disorders of amino acid metabolism, disorder of glucose metabolism, cystic fibrosis); and toxic insult (parenteral nutrition, drugs).
Infants with biliary atresia are typically born full term with normal birthweight. Shortly after birth, these infants present with persistent jaundice, pale stools, and dark urine. All premature and term infants who remain jaundiced after 21 and 14 days, respectively, should have serologic laboratory tests to evaluate potential liver disease. The cardinal biochemical feature of biliary atresia is conjugated hyperbilirubinemia. However, there is considerable overlap in the clinical, biochemical, radiologic, and histologic features of biliary atresia and other causes of neonatal jaundice on the differential. Radiologic tests are important in making an accurate and timely diagnosis, essential for timely surgical intervention. Prompt intervention is recommended as operative success is highest before 60 days, and efficacy declines as the infant ages.48
Diagnostic Tests
Initial diagnostic testing includes laboratory studies to identify cholestatic liver disease5 (Figure 41-2). Of note, serum γ-glutamyltransferase (GGT) is usually higher in biliary atresia than in other causes of neonatal cholestasis, especially when correlated with age.49
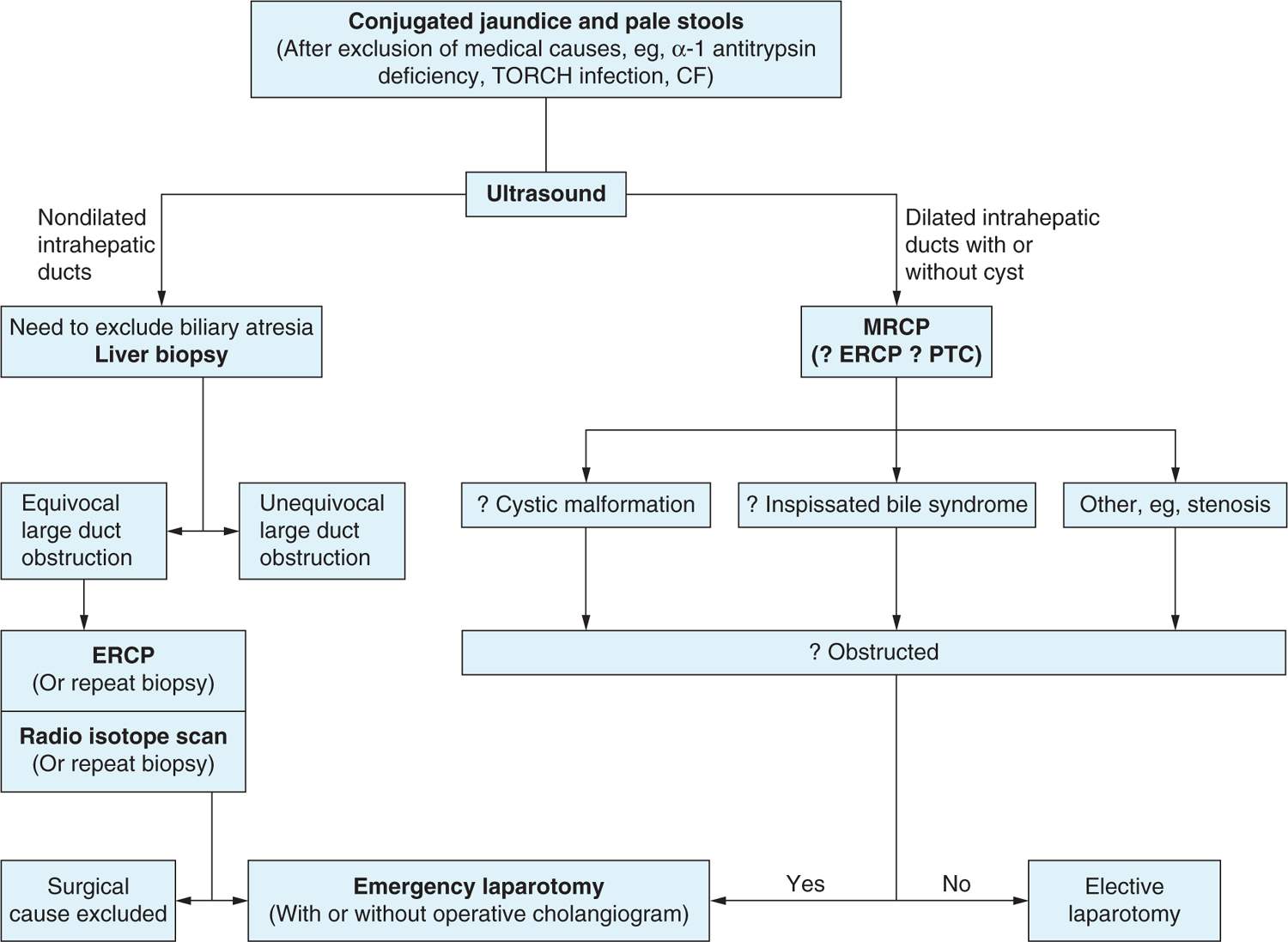
FIGURE 41-2 Suggested algorithm for investigation of infants. CF, cystic fibrosis; ERCP, endoscopic retrograde cholangiopancreatography; MRCP, magnetic resonance retrograde cholangiopancreatography; PTC, percutaneous transhepatic cholangiography; TORCH, toxoplasmosis, rubella, cytomegalovirus, herpes simplex virus. (Reprinted from The Lancet, Hartley, Davenport, and Kelly.5)
Ultrasound is the first step in imaging and is useful for excluding other causes of neonatal jaundice. An abdominal ultrasound will show an enlarged liver, absence of biliary dilation due to the inflammatory process, an absent or contracted gallbladder after a 4-hour fast,50 or a triangular cord sign (triangular or band-like echogenic density seen just above the porta hepatis).51 The sensitivity and specificity of a small or absent gallbladder in detecting biliary atresia range from 73% to 100% and 67% to 100%, respectively, when combined with clinical, pathologic, and subsequent surgical evaluations. The triangular cord sign is highly suggestive of biliary atresia52,53 but is operatordependent, with reported sensitivities varying from 49% to 73%54 to 83%–100% and with specificity of 98%–100%.55
Hepatobiliary scintigraphy (hepatobiliary iminodiacetic acid [HIDA] scan) can help distinguish biliary atresia from other etiologies of conjugated hyperbilirubinemia that do not need early surgical intervention56,57 by demonstrating the failure of bile excretion into the bowel. The reported sensitivity and specificity are 100% and 40%–100%, respectively.48 Improved specificity and sensitivity have been reported if the patient is premedicated with phenobarbital 5 mg/kg for 5 days.48 However, the specificity limits this radiographic tool in diagnosing biliary atresia.
Liver histology obtained on percutaneous biopsy is often considered the gold standard for the diagnosis of biliary atresia. One group reported accuracy of 96%–98% if the specimen contained at least 5–7 portal spaces,58 while another review reported that biliary atresia was only diagnosed correctly in 50%–99% of cases.55 Typical pathologic findings are extrahepatic biliary obstruction by varying degrees of portal tract fibrosis, edema, ductular proliferation, and cholestasis with the appearance of bile plugs. The possible presence of giant cell transformation may make the differentiation from other causes of neonatal hepatitis difficult. Of note, liver biopsy samples taken before 6 weeks of age, in the early development of biliary atresia, might not have the typical histologic features necessary for diagnosis.59
When the diagnosis is unclear, endoscopic retrograde cholangiopancreatography (ERCP) to visualize the biliary tract may be useful. Data supporting ERCP are that it is technically possible in 90% of infants and gives few false positives.60 However, it is a technically challenging procedure in neonates and generally not an option unless in a large center. Also, data supporting the use of ERCP for the diagnosis of biliary atresia are sparse. Magnetic resonance retrograde cholangiopancreatography (MRCP) has been evaluated as another potentially helpful tool for diagnosing biliary atresia, and results from initial studies are promising. In 2 recent studies, the negative and positive predictive values of MRCP detecting biliary atresia were from 91% to 100% and from 75% to 96%, respectively.61,62 The challenge with MRCP is the technical constraints in identification of luminal patency of the infantile bile ducts, which is often only 1 mm diameter or less.63 However, this noninvasive test may become more useful as it becomes more commonplace for radiologists and as the technology improves.
The true diagnostic gold standard is the intraoperative cholangiogram, done laparoscopically if there is doubt regarding the diagnosis before proceeding with Kasai portoenterostomy. If the diagnosis is made at the time of the cholangiogram, the surgeon can then proceed with the portoenterostomy.
Management
Portoenterostomy and liver transplantation remain the cornerstones of treatment of biliary atresia. Portoenterostomy, or the Kasai portoenterostomy, named after the Japanese surgeon Morio Kasai, who described the technique in the 1950s, is a procedure in which the entire extrahepatic biliary tree is excised so that the porta hepatis is transected at the level of the liver capsule, and the ductules that remain are exposed. A jejunal Roux loop is then anastomosed to the cut surface, completing a reconstruction (Figure 41-3). If successful, any patent intrahepatic bile ducts will drain into the Roux limb, allowing relief of the biliary obstruction. If possible, the Kasai procedure should be performed before 60 days of age when its short-term success is 80%.2,11,64 Efficacy of the procedure is thought to drop with age. However, this drop in efficacy may be limited to the less-common subgroups of patients with biliary atresia, including those with BASM syndrome or cystic biliary atresia. Supporting this notion is a large series that reported that children older than 100 days with isolated biliary atresia still had a native liver survival rate of 45% at 5 years.5,65
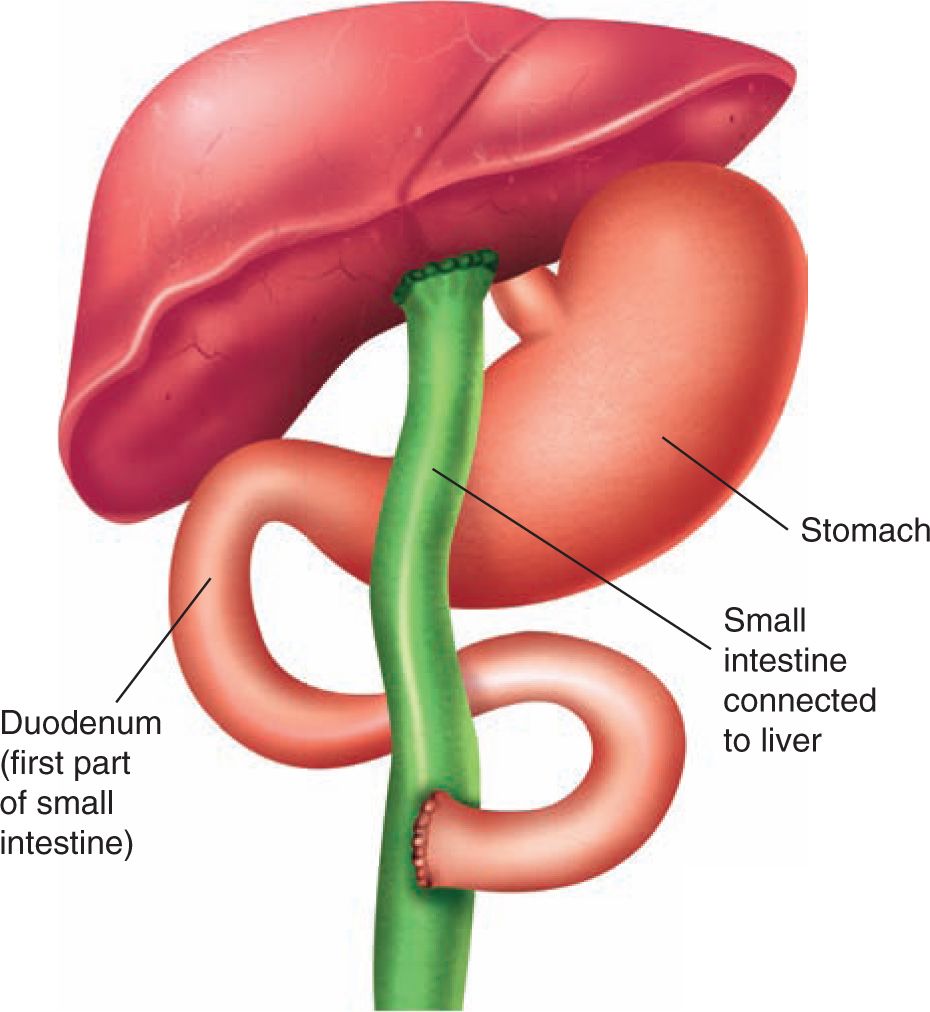
FIGURE 41-3 Kasai procedure. To perform the Kasai procedure, surgeons first carefully remove the damaged ducts outside the liver. They use a small segment of the patient’s own intestine to replace the ducts at the spot where bile is expected to drain. This segment not only connects to the liver but also connects to the rest of the intestine. The Y-shaped passageway formed by the Kasai procedure allows bile to flow from the liver into the intestine.
Because Kasai portoenterostomy is a palliative procedure and is not curative for biliary atresia, liver transplantation is the ultimate definitive therapy. Indications for liver transplantation depend on the success of the portoenterostomy and the rate of development of complications.5 In those infants who do not achieve adequate bile drainage with portoenterostomy, transplantation is usually indicated within 6 months to 2 years of age. In those children who had a successful portoenterostomy, liver transplantation is considered when the child has persistent or progressive cholestasis, development of cirrhosis with hepatic dysfunction, or development of portal hypertension with ascites and variceal bleeding unresponsive to endoscopic management. Factors that may predict the need for a liver transplantation are the bilirubin value 30 days postportoenterostomy66 and a score approaching 10 for pediatric end-stage liver disease assessment.67
In those children who have syndromic variants of biliary atresia rather than isolated biliary atresia, the associated anomalies increase the risk of early morbidity and mortality, creating a greater need for liver transplantation. Despite the increased potential for other anatomic anomalies, results are similar to those children with isolated biliary atresia.68
Outcome and Follow-up
Intrahepatic inflammatory processes will continue for at least 6 months after Kasai portoenterostomy.37 Therefore, even though the procedure may provide relief of mechanical obstruction and restoration of bile flow, subsequent fibrosis, cirrhosis, and portal hypertension will still ensue in the native liver.45,69 The rate of progression will vary, but progression is most likely in the setting of recurrent cholangitis.70 Actuarial survival with the native liver has been estimated at 32% to 61% at 5 years and 27% to 54% at 10 years of life.71–73 Factors that are most influential on long-term outcome are age (<60 days) and the size of the ductules (>150 μm) in the biliary remnants at the time of portoenterostomy.71,74,75
Other factors that contribute to long-term outcomes are episodes of cholangitis after surgery, the decade when surgery was performed, and the experience of the surgical team.72,76 In Japan, the actuarial survival of 307 patients treated with portoenterostomy improved71 from 20% before 1971 to 70% between 1971 and 1998. In the United Kingdom, a prospective study from 1993 to 1995 identified 93 cases of biliary atresia and showed that of 15 UK centers, only 2 operated on 5 or more cases per year, which resulted in a statistically significant difference in 5-year native liver survival (63% in centers with 5 or more cases and 14% in others) and overall survival.1 The higher success rates in larger-volume centers was confirmed by studies in France, which had improved survival rates.2
Those countries using living-related transplantation are reporting excellent outcomes, with 98% recipient 5-year survival.77 The largest follow-up study data from the United States reported 10-year actuarial graft survival of 73% and patient survival of 86% across 1976 patients.78 Transplantation also improves long-term catchup growth, nutrition, and maintenance of healthy development.79
ALAGILLE SYNDROME
Epidemiology
Alagille syndrome, first described in 1967, is an autosomal dominant genetic disorder affecting 1 in 30,000 live births,80 with variable expressivity of multisystem disease. The syndrome is characterized by a paucity of interlobular bile ducts with other associated features, including cholestasis (present in 96% of patients), cardiac anomalies (97%), butterfly vertebrae (51%), posterior embryotoxin of the eye (78%), and dysmorphic facies (96%).80 The dysmorphic facial features of AGS are described as a broad nasal bridge, triangular facies, and deep-set eyes. Renal anomalies, both functional and structural, and neurovascular accidents are much less common (15%) but can be associated with AGS.80
Pathophysiology
Microdeletion of the 20p12 gene corresponding to JAG1 is thought to result in AGS.81,82 The JAG1 gene is involved in signaling between adjacent cells during embryonic development, which influences how the cells are used to build organ systems in the developing embryo. Thus, mutations in JAG1 disrupt this signaling pathway, resulting in errors of development, especially of the heart, bile ducts in the liver, spinal column, and certain facial features.
When the bile ducts are narrowed and malformed, the bile produced in the liver is unable to be transported to the small intestine. Thus, the bile builds up in the liver and causes scarring and eventual cirrhosis.
Differential Diagnosis
As most infants present with cholestatic jaundice, 1 of the first differentials is biliary atresia. Other differentials considered include congenital hepatic fibrosis, cystic fibrosis, neonatal jaundice, polycystic kidney disease, progressive familial intrahepatic cholestasis, and tyrosinemia.
Diagnostic Testing
Clinical suspicion should drive the initial workup and evaluation of suspected AGS. Because AGS potentially involves many systems, ophthalmologic examination, echocardiogram, and imaging of the vertebrae are all useful in ascertaining the diagnosis. For those infants who present with cholestatic disease, initial evaluation should include liver function tests, prothrombin time, and levels of fat-soluble vitamins. In these patients, an ultrasound should also be considered to rule out biliary atresia. However, there are no ultrasound findings specific to AGS. Hepatobiliary scintigraphy, and intraoperative cholangiogram may also be useful in distinguishing AGS from biliary atresia or choledochal cysts. The challenge with a HIDA scan is that up to 61% of patients with AGS have been reported to have no evidence of tracer excretion from the liver, as would be expected with biliary atresia.83 In these patients, cholangiogram, either percutaneous or intraoperative, may be useful. If infants with AGS have hypoplasia of the extrahepatic biliary tree,83 liver biopsy can be useful for diagnosis.
The advent of genetic testing for AGS has led to an increase in diagnosis. Deletion or mutation of the JAG1 gene on the short arm of chromosome 20 is present in 60% to 70% of patients with AGS.84 Genetic testing should be conducted for any patient for whom there is suspicion of AGS.
Management
Management of AGS focuses on treatment of the individual components of the disease. Cholestasis is treated medically with choleretics, most commonly ursodiol. Debilitating pruritus from hyperbilirubinemia in AGS can be addressed with antihistamines, sedatives, and rifampin.85
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


