Cyanotic Congenital Heart Defects
INTRODUCTION
Cyanotic congenital heart defects are the most important group of defects necessitating surgical intervention in the neonatal period. In general, this group of defects is characterized by ductal dependency of pulmonary blood flow (or effective pulmonary blood flow) but with notable exceptions of truncus arteriosus (TrA) and obstructed total anomalous pulmonary venous return or connection (TAPVR).
Cyanotic heart defects should be considered and ruled out or confirmed in any newborn presenting with cyanosis (see Chapter 80 for differential diagnosis of cyanosis in the neonate), although in a cyanotic neonate the response to oxygen, heart murmurs, chest roentgenogram, and electrocardiogram (ECG) can offer clues to cardiac defects and prompt further evaluation. The echocardiogram is the mainstay of diagnosis to delineate the morphology of the heart defect and plan intervention.
In the current era, fetal obstetric ultrasound and fetal echocardiography have enabled us to accurately diagnose congenital heart defects in the majority of neonates before they are born.
This modality has changed the presentation and management of this group of neonates. Because the diagnosis is often established before birth, the emphasis has shifted more on planning the management immediately after birth rather than establishing the diagnosis as in the past. However, a neonatologist still is faced with undiagnosed cyanotic newborns for which all clinical skills can be tested.
Once the diagnosis is confirmed, adequacy of pulmonary blood flow and systemic blood flow should be assessed and pulmonary venous obstruction should be ruled out. Physiology-specific therapeutic medical management should be instituted, and surgical treatment should be planned. In patients with low birth weight or in premature neonates, the approach to “wait and grow the patient” has not been promising. Waiting prior to surgical palliation increases preoperative complications without gaining any benefits.1 Even in this group of patients, an early correction is indicated, and in patients with functional single ventricles, appropriate early surgical palliation is indicated.
TOTAL ANOMALOUS PULMONARY VENOUS RETURN
Introduction
Total anomalous pulmonary venous return (TAPVR) or connection is a cardiac malformation in which there is no direct connection between any pulmonary vein and the left atrium; rather, all the pulmonary veins connect to the right atrium or one of its tributaries. A patent foramen ovale (PFO) or atrial septal defect (ASD) is present and is necessary for survival after birth in all newborns with TAPVR.2
TAPVR is a rare congenital cardiac anomaly occurring in about 1.5% of children born with congenital heart defects. In addition, it can occur as an associated defect with many other congenital cardiac defects, especially in newborns with heterotaxy syndromes with single functional ventricles. Nevertheless, it is one of the few lesions necessitating urgent or emergent cardiac surgery in the neonatal period.
Morphology
TAPVR morphology (Figure 18-1A) can be categorized into 4 broad anatomic types:
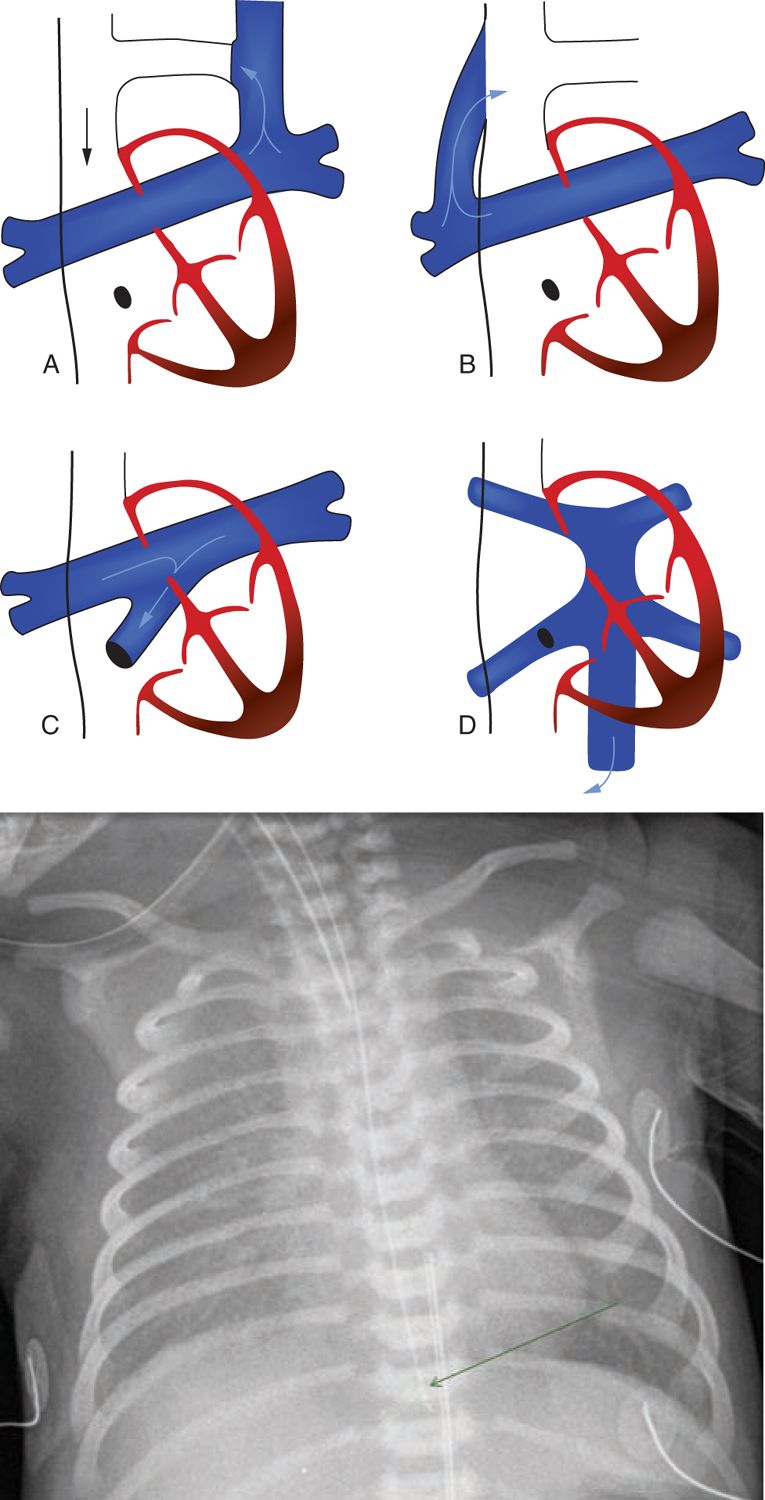
FIGURE 18-1 A, Types of total anomalous pulmonary venous return (TAPVR): a and b, supracardiac; c, cardiac; d, infracardiac. (From Stark et al.58) B, Chest x-ay of 1-day-old with obstructed infracardiac total anomalous Pulmonary venous connection, showing a ground-glass appearance in bilateral lung fields with a small cardiac shadow.
1. The supracardiac type is the most common (45%); here, the pulmonary veins drain via either the ascending vertical vein to the innominate vein or directly to the superior vena cava (SVC) and rarely to the azygos vein. About two-thirds of these have pulmonary venous obstruction, often at the junction of the vertical vein to the systemic vein.
2. In the cardiac type (20%), drainage is directly into the right atrium or into the coronary sinus (CS). Pulmonary venous obstruction is rare but may be seen in up to 20% at the CS ostia or orifices of individual veins or their confluence.
3. In the infracardiac type (25%), drainage occurs via the descending vertical vein into the portal vein or rarely into the inferior vena cava. These are by definition always obstructed to some extent because the pulmonary venous return has to pass through the hepatic capillary system. In addition, they have a high incidence (80% to 90%) of obstruction, often at the junction of the vertical vein to the systemic vein.
4. The mixed type (the least common at 5%–10%) consists of a combination of one or more of the 3 other types described. It can be technically most challenging to repair.
In patients with isolated TAPVR, atrial septal communication, which is essential for survival, and a patent ductus arteriosus (PDA) are the only associated defects. The cardiac chambers and valves are almost always normally formed.
Even the youngest infants dying with the malformation often have structural changes in the lungs. Increased pulmonary arterial (PA) muscularity is seen in all infants dying with TAPVR, as evidenced by arterial wall thickness and vein wall thickness. There is also extension of muscle into smaller and more peripheral arteries than normal.
Clinical Presentation, Pathophysiology, and Diagnosis
Neonates usually present with tachypnea and respiratory distress.3 Cyanosis is prominent when there is pulmonary venous obstruction. In the absence of obstruction, cyanosis is often overshadowed by tachypnea. Right-to-left shunting at the level of the PDA also adds to the cyanosis.
The neonate with obstructed TAPVR often is in critical condition, requiring tracheal intubation and ventilator support. It is not uncommon to see these patients mistakenly diagnosed as having pulmonary pathology such as respiratory distress syndrome, meconium aspiration, bronchiolitis, and so on. TAPVR should always be considered in the differential diagnosis of any neonate presenting with cyanosis or tachypnea.
In TAPVR, the right atrium is the common mixing chamber, which often is reflected in the frequent finding of the close similarity of oxygen content and saturations from the right atrium, left atrium, PA, and systemic artery. However, because of streaming of systemic venous return in the right atrium, directing inferior vena caval blood through the foramen ovale to the mitral valve and SVC blood through the tricuspid valve (TV), there can be considerable variation in different patients. This can be affected further by the size of the atrial communication. The hemodynamics of the nonobstructive types of TAPVR is similar to that of a large ASD. These neonates may present later in the neonatal period as the right ventricular (RV) compliance increases and the pulmonary vascular resistance (PVR) falls, and the ASD may become relatively inadequate in size.
A chest x-ray (Figure 18-1B) of neonates with TAPVR and pulmonary venous obstruction shows diffuse haziness or, in severe forms, a ground-glass appearance. In the supracardiac type, the appearance often is described as a “snowman sign.” These signs are less marked when the pulmonary circuit can decompress via a PDA.
In the current era, even with fetal screening, TAPVR is often undiagnosed unless a fetal cardiac echocardiogram is part of prenatal evaluation. Postnatally, cardiac echo (Figure 18-2) is the definitive means of diagnosis. In some, a cardiac catheterization and angiography or a high-resolution computed tomographic (CT) scan may be needed to define the pulmonary venous anatomy.
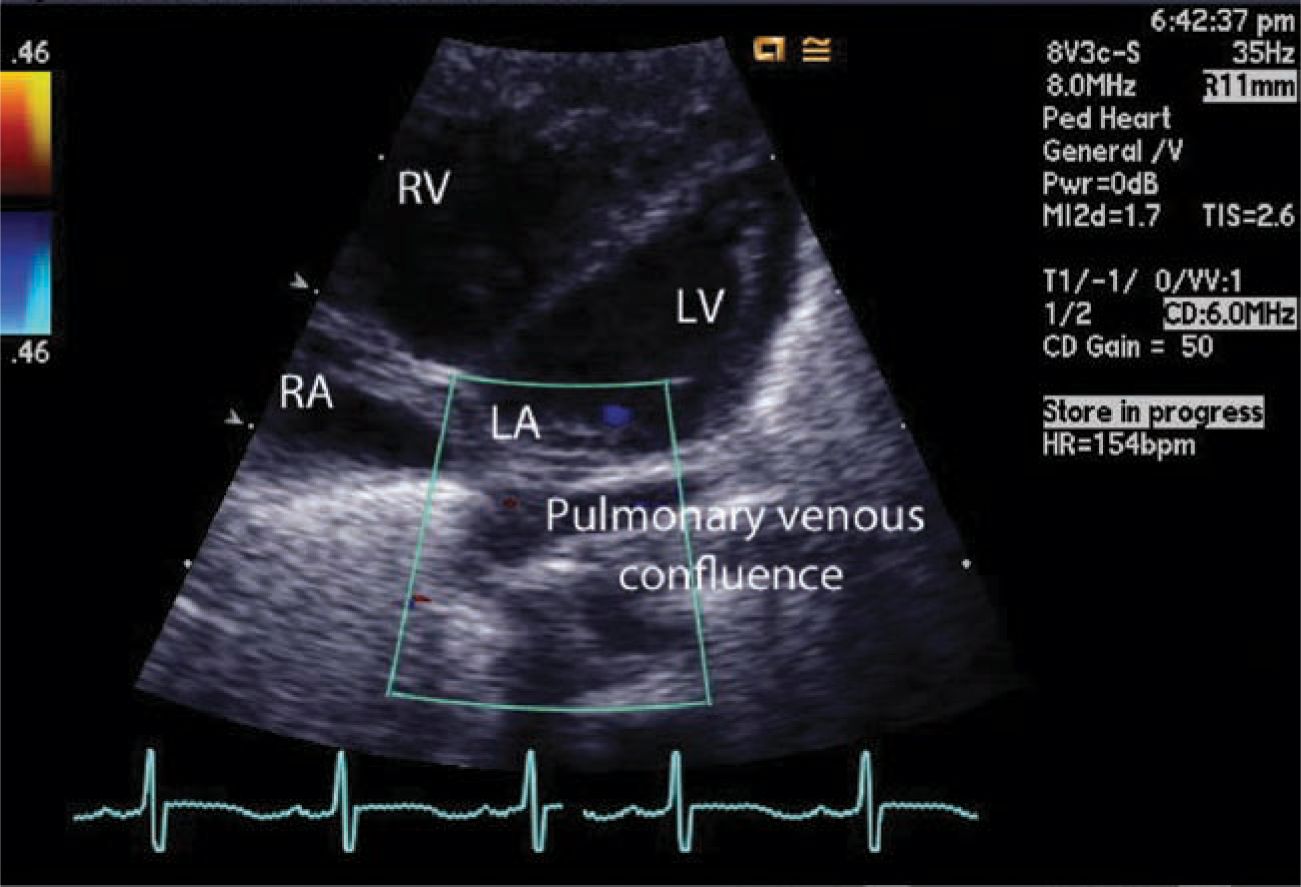
FIGURE 18-2 Echocardiography. Pulmonary venous confluence below the small left atrium pulmonary veins does not connect to the left atrium. Because of the pulmonary hypertension, the right ventricle is enlarged and the left ventricle is relatively small because of underfilling.
Neonates born with TAPVR in general have an unfavorable prognosis. Mortality during the first few weeks or months of life is common in most neonates in whom tachypnea, cyanosis, and clinical evidence of low cardiac output develop, with only about 50% surviving beyond 3 months of life. Such neonates usually have pulmonary venous obstruction, long pulmonary venous pathways, and a small interatrial communication. Only about half the neonates surviving to age 3 months survive to 1 year.
Management
Regarding management,4–6 surgery should be undertaken emergently, often immediately after diagnosis, in neonates and infants who are critically ill. These are usually neonates with obstructed TAPVR. A brief period of preoperative preparation and stabilization while the diagnosis is being confirmed is all that should be expected. This involves correcting metabolic acidosis and optimizing the ventilation. Prostaglandin E (PGE) is not indicated unless suprasystemic RV pressure is documented by echo. In such instances, right-to-left ductal shunting may improve systemic cardiac output. Rarely, the neonate is so critically ill with significant pulmonary injury that extracorporeal membrane oxygenation (ECMO) may be indicated for a few days prior to surgery. However, in stable neonates, typically without pulmonary venous obstruction, the operation can be scheduled electively.
Surgery is performed on cardiopulmonary bypass with or without circulatory arrest. This commonly involves anastomosing the common pulmonary venous chamber to the back of the left atrium (Figure 18-3). In TAPVR to the CS, the CS is flayed open into the left atrium. If the pulmonary veins are stenotic, each individual vein is flayed open; currently, a suture-less technique is favored in this group. This involves suturing the margins of the cut-open back wall of the left atrium to the pericardium beyond the pulmonary venous openings.
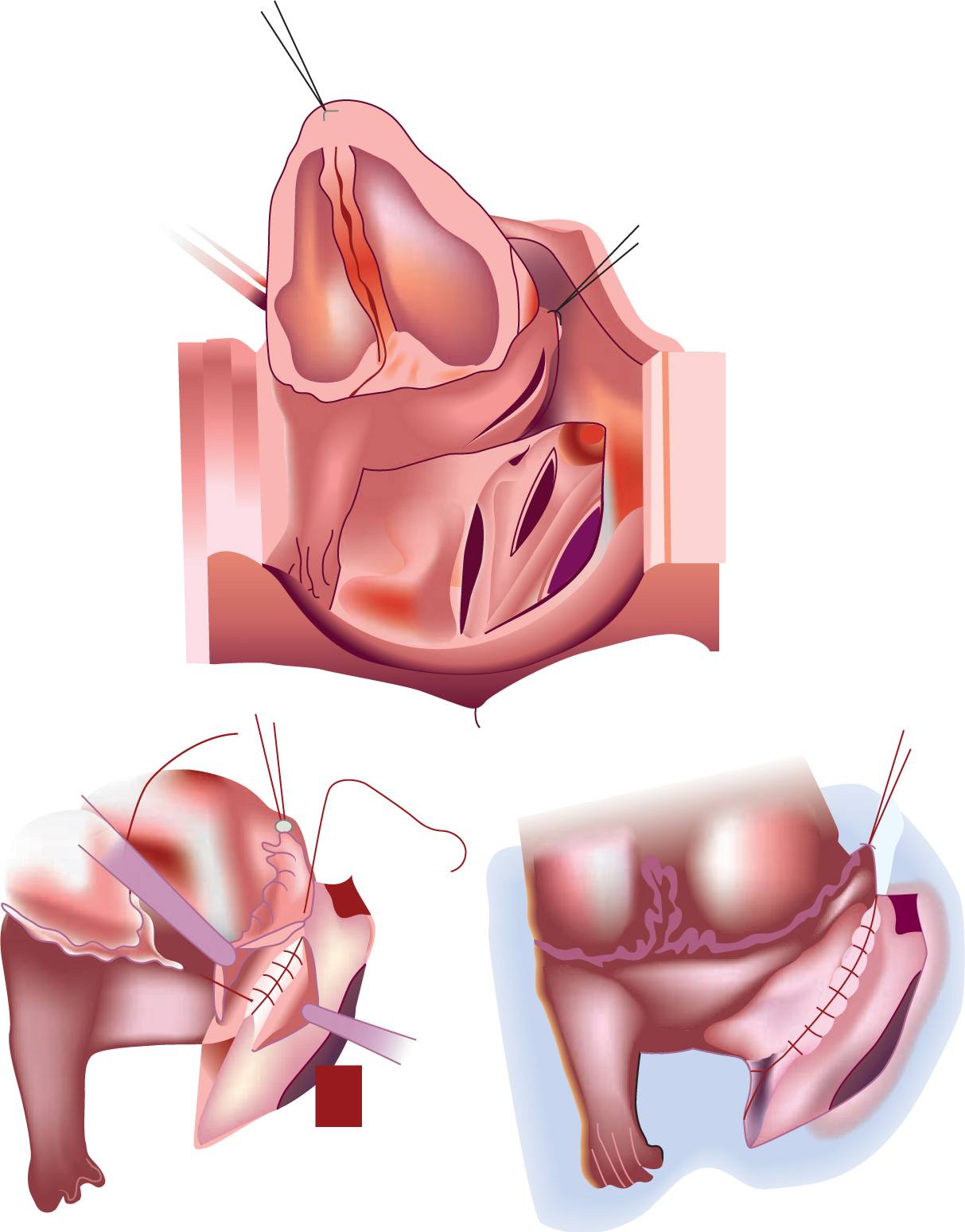
FIGURE 18-3 Repair of infracardiac-type total anomalous pulmonary venous drainage. Pulmonary venous chamber is opened and anastomosed into the back of the left atrium. (From Khonsari et al.59)
Postoperative management can be challenging in these neonates. They often have high PVR, which will require aggressive treatment of acidosis, hyperventilation, and inhaled nitric oxide. The left ventricle (LV) is also often noncompliant and is sensitive to preload. Monitoring right atrial, left atrial, and PA pressure with indwelling catheters placed during surgery is helpful.
Outcomes
Surgical outcome7–9 in neonates has steadily improved in the decades since 1970. Data from the database of the Society of Thoracic Surgeons show overall early mortality was 12.0% between 2002 and 2004. For infants falling within the normal weight range, mortality was 9.9%; it was 29.2% for infants less than 2.5 kg body weight.
Recurrent pulmonary venous obstruction is seen in 5% to 10% of neonates and often occurs within a few weeks to months after surgery. While anastomotic stenosis is uncommon and can easily be corrected by reoperation, true pulmonary vein obstruction is often not amenable to surgical or any other form of intervention.
TRANSPOSITION OF GREAT ARTERIES
Introduction
Complete transposition of the great arteries (TGA) is probably the most common cyanotic congenital heart defect in neonates. It accounts for approximately 5% of all congenital heart defects with male preponderance. If untreated (without PGE or balloon septostomy), mortality is high: 30% at 1 week, 50% at 1 month, and 90% at 1 year.
Morphology
Morphologically,10,11 in TGA, as the name implies, the aorta is transposed to the RV and the PA to the LV. The aorta is anterior and commonly to the right of the PA; therefore, the prefix d is used for dextroposition, which is the most common type of complete transposition (d-TGA). The atria and ventricles are in normal relationship. Although the coronary arteries arise from the aorta, in at least a third the coronary artery origins and course are abnormal. All patients with TGA have an interatrial septal communication (PFO or ASD) and a PDA at birth. In about 70% of patients with d-TGA, the ventricular septum is intact (d-TGA/IVS), and the remaining 30% have a ventricular septal defect (d-TGA/VSD). Pulmonary stenosis, coarctation of the aorta (COA), and aortic arch obstruction are seen in about 25% to 35% of patients with d-TGA/VSD but they are rare in patients with d-TGA/IVS.
Coronary arteries originate from the sinuses of Valsalva facing the PA and are normal in two-thirds of the patients, with the left coronary artery originating from the leftward anterior-facing sinus and the right coronary artery (RCA) from the rightward posterior-facing sinus. In the rest, the pattern of origin and course is abnormal. The most common is the origin of the circumflex coronary artery from the RCA. A single origin of all coronary arteries and an intramural course of the coronary artery within the aortic wall are not infrequent and can pose technical challenges for the surgeon.
Pathophysiology
In d-TGA, the systemic venous blood returning to the RV is ejected back into the systemic bed through the aorta without passing through the lungs, and the pulmonary venous blood returning to the LV is ejected back into the lungs through the PA. This arrangement of 2 parallel circulations does not affect fetal survival because the placenta is the source of oxygenated blood but is incompatible with life in the postnatal period. Hence, after birth survival depends on adequate communication between the 2 parallel circuits at the level of PDA, ASD, and VSD (if present). The extent of mixing is determined by vascular resistance in each of the circulations, size of the communications between the 2 circuits, compliance of the ventricles, and volume of blood flow. Inadequate mixing results in hypoxia, hypercapnia, metabolic acidosis, and eventually death if untreated. Immediate postnatal interventions are primarily designed to maximize mixing of oxygenated and deoxygenated blood.
Clinical Presentation and Diagnosis
Cyanosis is the dominant clinical feature of these neonates, and it is often unresponsive to inhaled oxygen. Cyanosis may be less apparent in neonates with large ASDs and large PDAs, except occasionally because of streaming there is inadequate mixing of oxygenated and deoxygenated blood resulting in intractable cyanosis. In neonates with d-TGA/VSD, cyanosis is less apparent.
At presentation, these neonates often have unstable hemodynamics and a varying degree of acidosis. Physical examination may reveal a mildly hyperactive precordium with prominent single second heart sound and soft systolic murmurs. Peripheral arterial pulses may be prominent if the PDA is large. This may also result in symptoms of congested lungs. Chest x-ray often shows a mildly enlarged heart with a typical appearance (Figure 18-4).
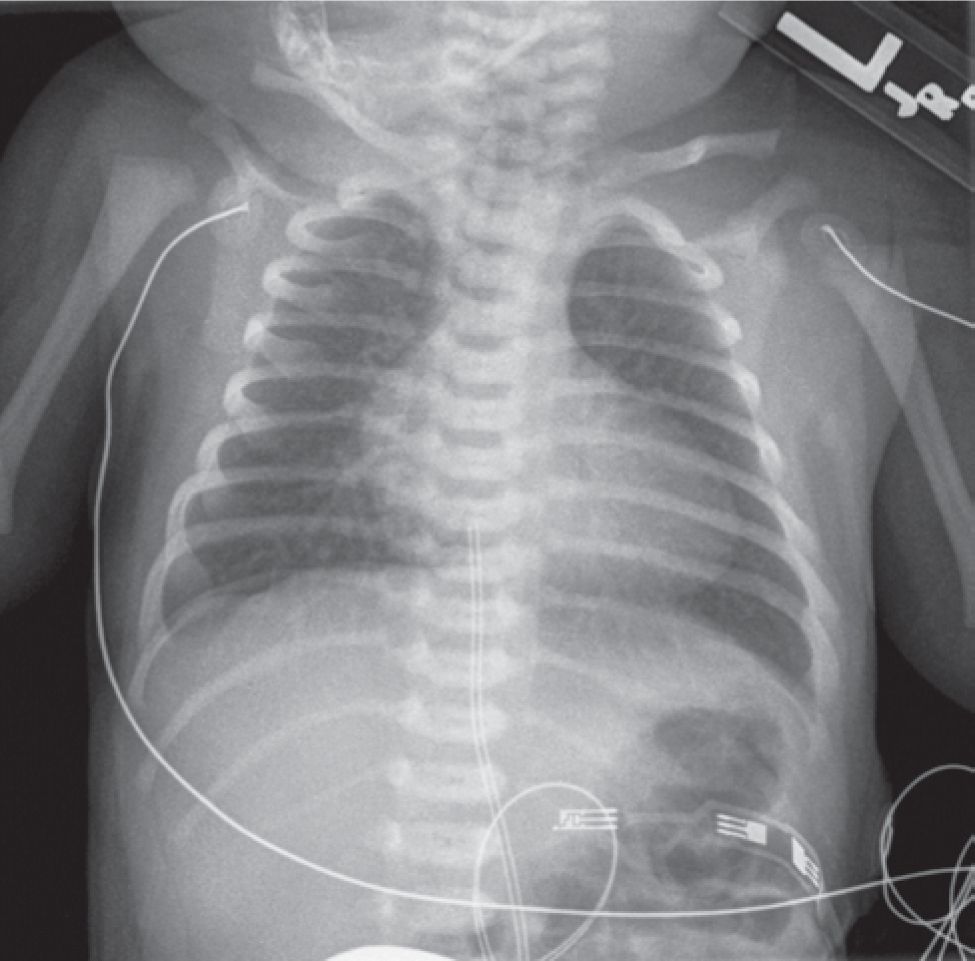
FIGURE 18-4 Chest x-ray. The heart is enlarged with a narrow pedicle, showing an “egg-on-a-string” sign.
Depending on the institution and country, in the current era prenatal diagnosis of TGA varies from 20% to 75%, and in such instances planned management almost always results in a stable neonate. Echocardiography is the mainstay of diagnosis and most often the only modality of imaging needed (Figure 18-5). Echocardiography can give all the necessary information, including the coronary anatomy needed for surgical planning. Rarely, a CT angiogram or a cardiac catheterization is needed.
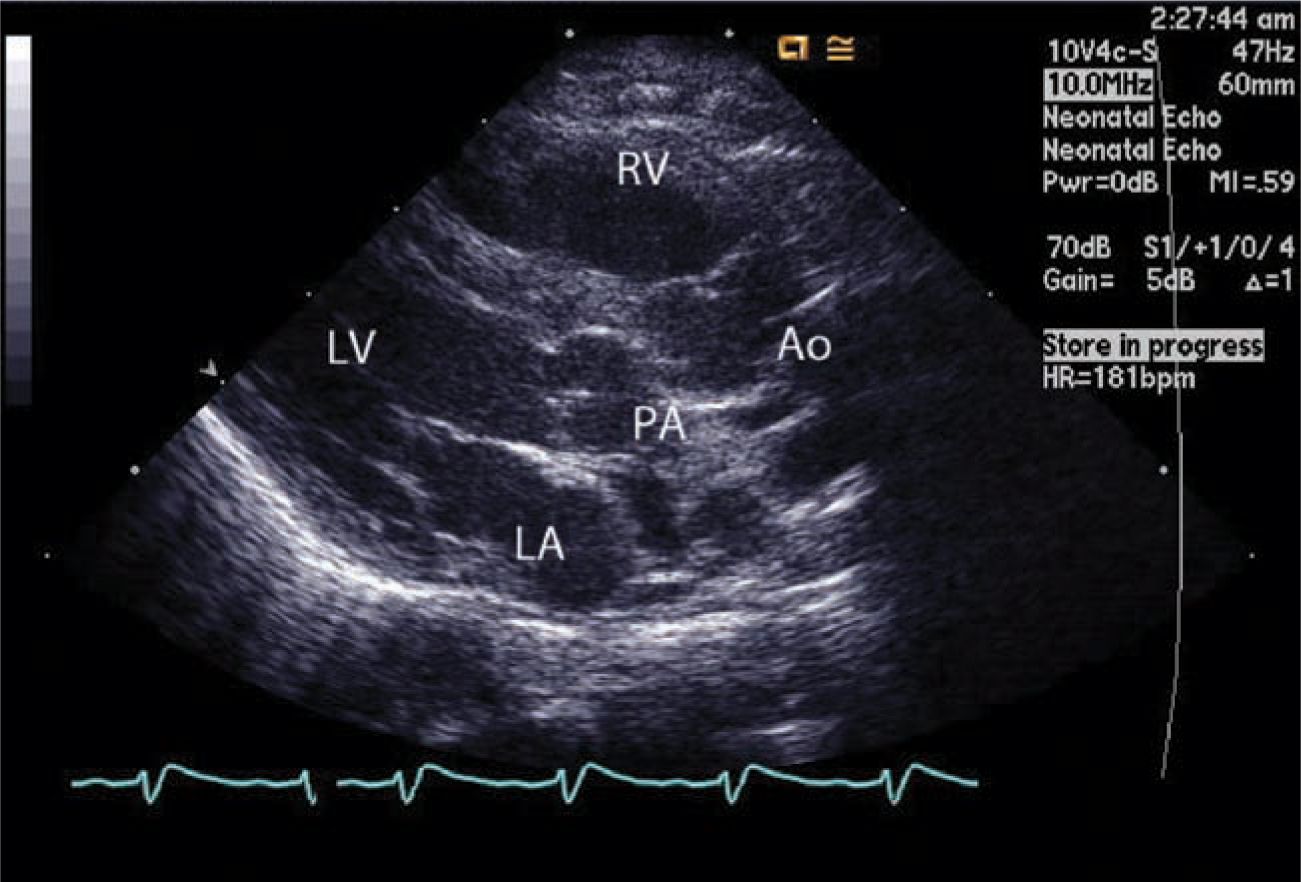
FIGURE 18-5 Echocardiography of the transposition of the great arteries. The aorta (Ao) arises from the right ventricle (RV) anteriorly, whereas the pulmonary trunk arises from the left ventricle (LV) posteriorly. LA, left atrium; PA, pulmonary artery.
Cardiac catheterization is usually a palliative tool in TGA to perform balloon septostomy.
Management
The preoperative management of a neonate with TGA/IVS is directed to increase mixing of oxygenated and deoxygenated blood between the 2 parallel circulations with the goal of increasing systemic oxygen delivery. PGE2 infusion is started, and if necessary, a balloon atrial septostomy is performed either at the bedside in the neonatal intensive care unit (NICU) under echo guidance or in the cardiac catheterization lab. Most neonates do not require mechanical ventilation.
Some patients may not have adequate mixing of the oxygenated and deoxygenated blood despite these interventions; in such patients, inotropic support to increase the cardiac output and paralyzing the neonate and providing mechanical ventilation to decrease the metabolic needs may be necessary. Rarely, emergent surgery may be needed. In addition to these measures, correcting the metabolic abnormalities may be necessary.
On the other hand, neonates with TGA/VSD generally do not require PGE2 or balloon septostomy if the VSD is large.
Surgical treatment is undertaken within the first few days of life, and it is not advisable to delay beyond 2 weeks of birth.12 If there is a large VSD, some surgeons elect to wait for 2 to 4 weeks. The current procedure of choice is the arterial switch operation (ASO; Figure 18-6), which has replaced the atrial switch procedures, such as the Senning and Mustard operations.
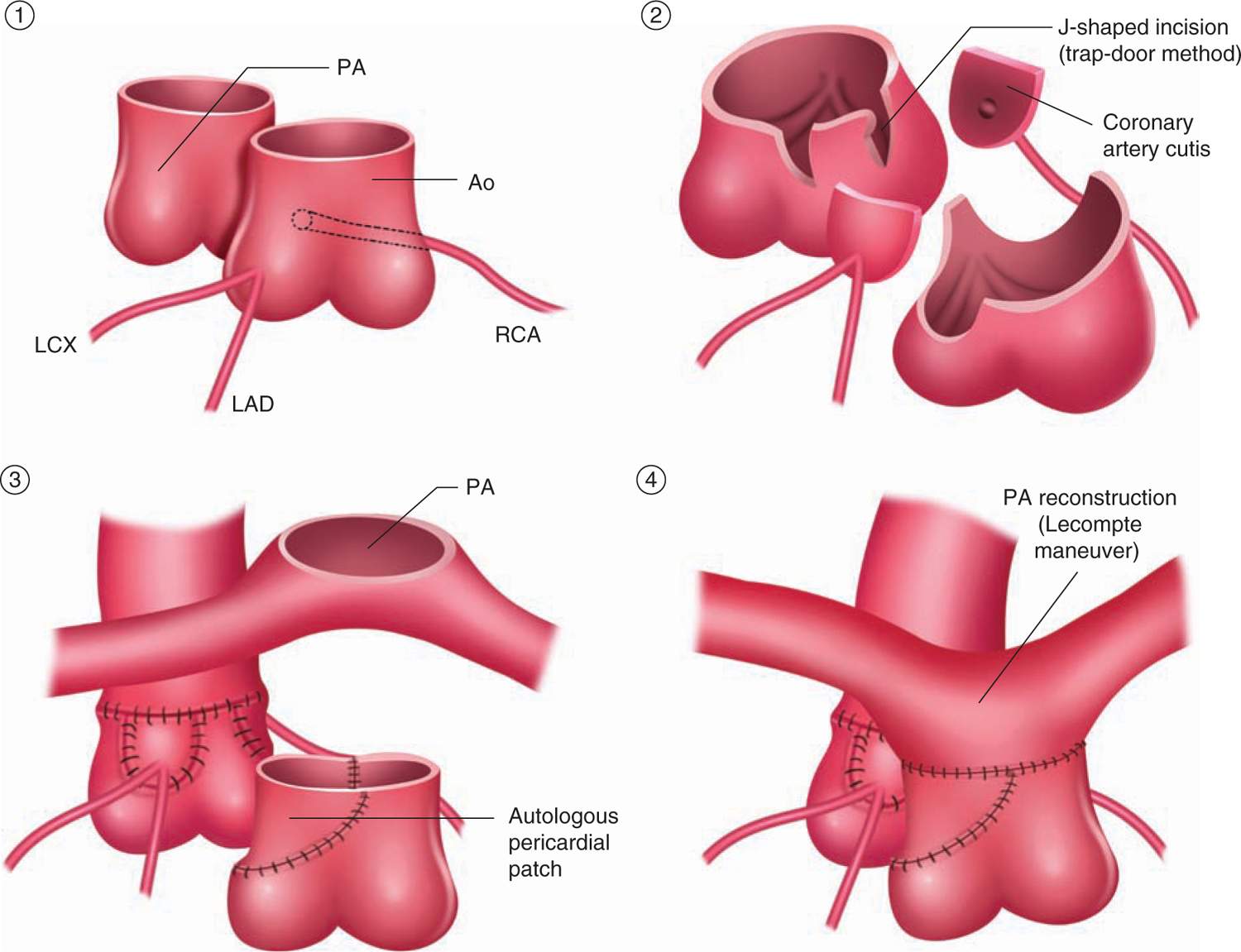
FIGURE 18-6 Surgical repair of arterial switch operation. Two coronary arteries are harvested and transferred into neoaorta. The pulmonary artery is usually moved anterior to the aorta (Lecompte maneuver to prevent obstruction). LCx, left circumflex branch; LAD, left anterior descending branch; RCA, right coronary artery. (From Yasui et al.60)
The ASO is performed with cardiopulmonary bypass under cardiac arrest with cardioplegia with or without hypothermia. The procedure consists of transferring the aorta and coronary arteries to the LV and the PA with its branches to the RV.
In this procedure, the native pulmonary valve becomes the neoaortic valve and the native aortic valve becomes the neopulmonary valve. In addition, the PAs are translocated anterior to the aorta (Lecompte maneuver). Transferring the coronary arteries, the neoaorta13 is the critical step in ASO. The PDA is ligated and divided. The ASD and VSD, if present, are closed.
Associated defects like COA or aortic arch obstruction are also repaired. In patients with LV outflow tract obstruction (LVOTO), an assessment is made whether it is dynamic or if it can be resected. In some patients with TGA/VSD/LVOTO, the pulmonary annulus is hypoplastic, the subpulmonary LV outflow tract (LVOT) is hypoplastic, or both. In such instances, a palliative procedure such as a systemic-to-PA shunt is undertaken; a primary repair is preferred by some surgeons. The operations for this subset of patients are known as the Rastelli procedure, REV procedure, or Bex-Nikkaidoh procedure. In the Rastelli or REV procedures, the LV is baffled to the aorta through the VSD; in the Bex-Nikkaidoh procedure, the aorta along with the valve and coronaries are translocated to the LV.
Outcomes
The early results12,14–17 for the ASO have significantly improved since the late 1980s. The current survival rate for the ASO in those with TGA with or without VSD is less than 5%. If one excludes the high-risk patients, then the mortality is about 1% or less in most major centers. The risk factors for early death are also variable with experience. However, prematurity, low birth weight, single intramural coronary artery, aortic arch obstruction, multiple VSDs, hypoplasia of the RV, and older neonate/infant with TGA/IVS are some important risk factors. Many of these are neutralized in experienced hands. Preoperative physiologic state is also an important contributor to early death.
Late supravalvar PA stenosis and coronary artery obstruction are uncommon but important complications that require continued follow-up in these infants.
Another rarer complication, neoaortic valve regurgitation, may require even longer follow-up to determine its extent and significance.
TETRALOGY OF FALLOT
Introduction
Tetralogy of Fallot (TOF) is a common cyanotic congenital heart defect and accounts for 10% of all congenital heart defects. In 1888, Fallot described 4 related cardiac abnormalities, which together are known as TOF: VSD; PA stenosis; dextrorotation of the aorta (rightward), which overrides the VSD; and RV hypertrophy. Untreated, about 30% do not survive beyond 3 months, and 10% survive to the age of 21.
Morphology
Morphologically,18,19 most likely the anterocephalad deviation of the outlet septum and an abnormal relationship of this outlet septum to the rest of the interventricular septum result in all 4 morphologic features. The VSD in TOF is typically conoventricular or perimembranous, but it can also be subarterial with an absent outlet septum. In about 5% of patients, an additional VSD may be present. The crowding of the RV outflow tract (RVOT) causes RV outflow tract obstruction (RVOTO). This results in varying degrees of pulmonary valve abnormality (stenosis, hypoplasia, or atresia) and sometimes supravalvar PA narrowing. The subvalvar infundibulum is narrow and muscle bound in a majority of patients. In a small number of patients, the PAs can be hypoplastic. In the subgroup of patients with pulmonary valve atresia, the PAs are typically duct dependent. In about 25% of these patients, the pulmonary blood flow is derived from aortopulmonary collaterals.
In about 5% of patients with TOF, the coronary artery abnormalities are the most common and are the origin of the left anterior descending (LAD) branch from the RCA.
Clinical Presentation, Pathophysiology, and Diagnosis
Because of RVOTO, the venous blood from the RV is ejected into the aorta through the VSD; this is facilitated to some extent by the overriding of the aorta into the RV. Cyanosis becomes more pronounced as the severity of RVOTO increases. In patients with pulmonary valve atresia, all the venous blood is ejected into the aorta. As the severity of RVOTO increases, so does the dependency of pulmonary blood flow on the presence of a PDA or aortopulmonary collaterals.
Most neonates with TOF are asymptomatic at birth, and cyanosis may be the only sign; this is often undetected if there is a PDA or in milder forms of TOF. In neonates with significant RVOTO, the cyanosis is obvious. In those with severe RVOTO or pulmonary valve atresia, the pulmonary blood flow is dependent on the patency of the ductus arteriosus. The neonates quickly become hypoxic as the PDA starts to close and require PGE2 infusion to maintain systemic oxygenation.
Cyanotic spells are episodes of severe hypoxia and can be seen in newborns with moderate-to-severe RVOTO. The exact mechanism of these spells is unclear but may be multifactorial; the decrease in systemic vascular resistance and infundibular muscular hyperactivity are thought to be important.
Most neonates with TOF who have well-balanced systemic-to-pulmonary blood flow and systemic arterial oxygen saturation above 85% are discharged and have elective surgery between 2 and 6 months of age. The neonates who are on PGE2 and who have cyanotic spells require surgical intervention in the neonatal period.
Clinical examination often reveals a systolic ejection murmur and a soft second heart sound. A continuous murmur of a PDA may be present. Chest x-ray (Figure 18-7) and ECG are often not diagnostic in neonates. Echocardiography (Figures 18-8 and 18-9) is the mainstay of diagnosis. With this modality, the diagnosis can be accurately established and all other morphologic features clearly defined except the details of pulmonary blood supply in patients with hypoplastic PAs or those with pulmonary atresia. In these neonates, cardiac catheterization and angiography or CT angiography is indicated.
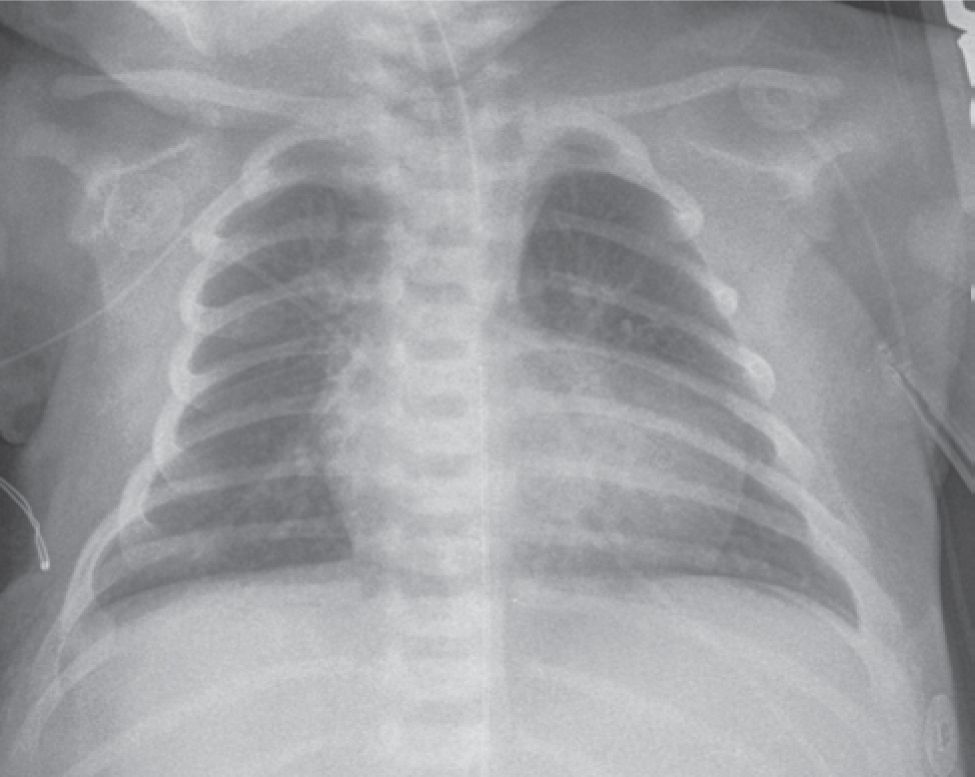
FIGURE 18-7 Chest x-ray of 1-month-old with tetralogy of Fallot (TOF) with severe pulmonary stenosis. Because of the diminutive pulmonary artery, a characteristic pattern, called the couer en Sabot “boot-shaped heart” is seen. The pulmonary vascular bed is hypoperfused.
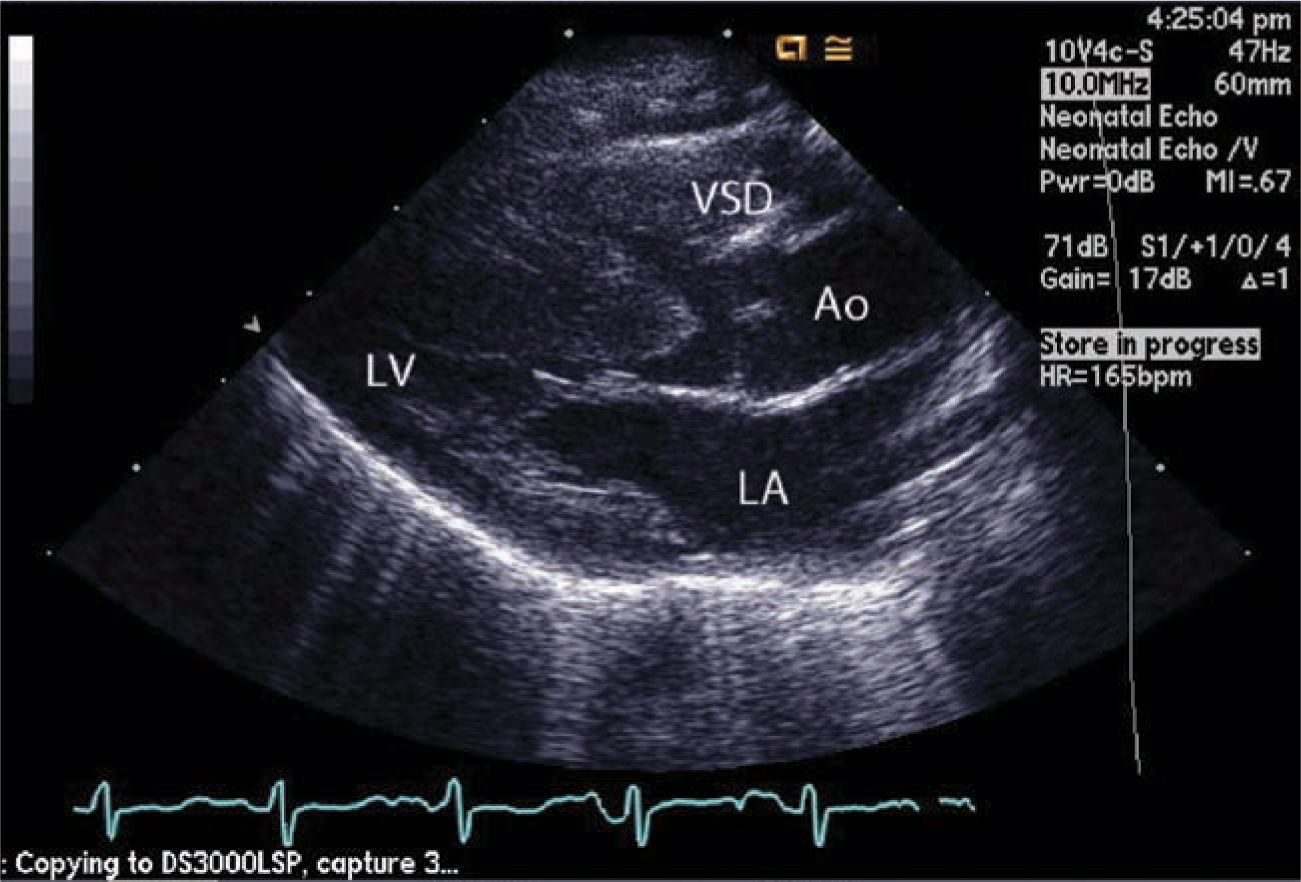
FIGURE 18-8 Echocardiography. Aorta (Ao) is overriding significantly to the right ventricle (RV). Because of pulmonary artery stenosis, the shunt through the ventricular septal defect (VSD) is right to left. LA, left atrium; LV, left ventricle.
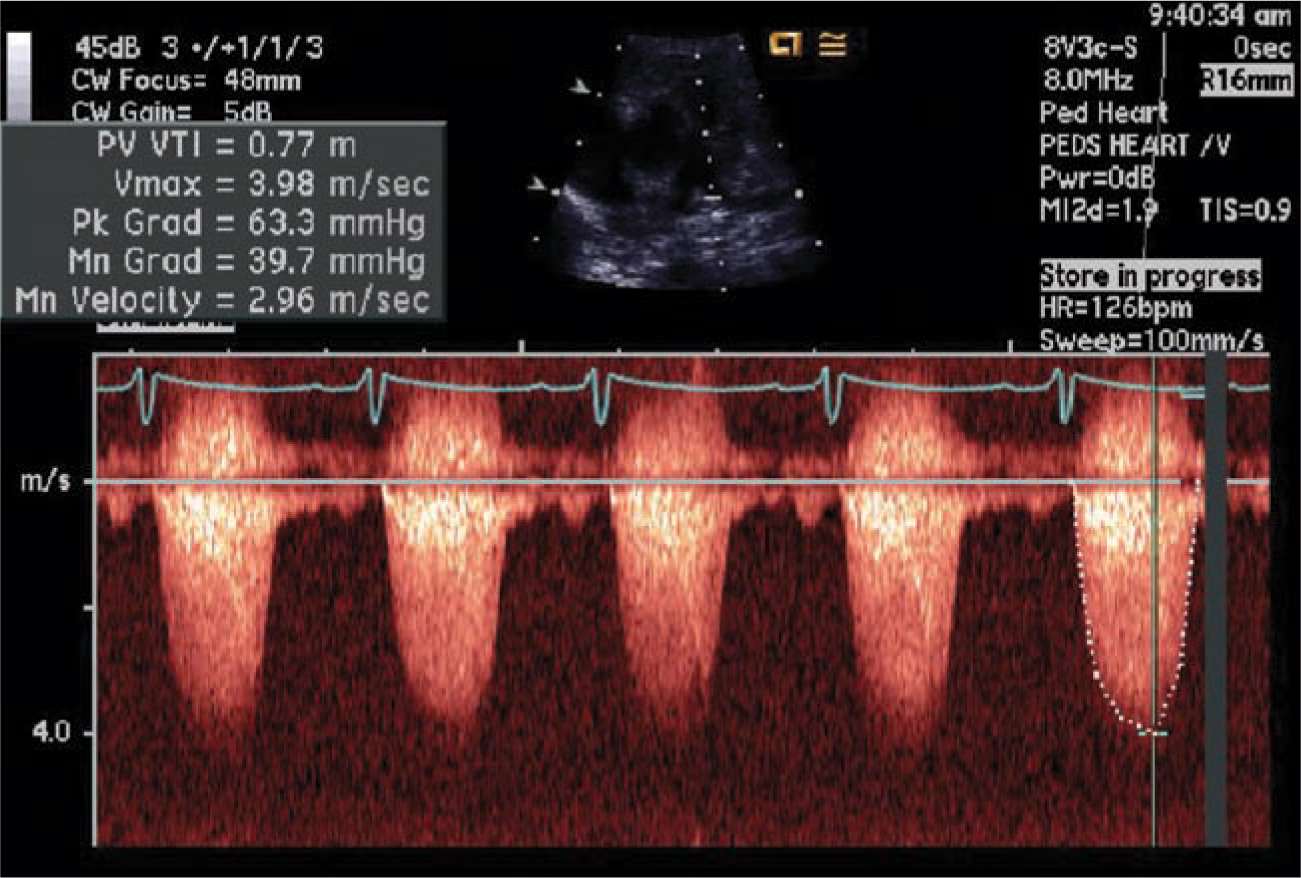
FIGURE 18-9 Doppler echocardiography of obstructed right ventricular outflow tract (RVOT) in a neonate with tetralogy of Fallot (TOF). The flow across the RVOT is accelerated significantly (Vmax 3.98 m/s).
Management
Most neonates with TOF do not require any intervention other than a period of observation until the PDA closes to ensure that they have adequate antegrade pulmonary blood flow to maintain systemic arterial oxygen saturation (preferably above 85% on room air).
If the neonate has cyanotic spells, aggressive medical management is immediately indicated. These spells are often amenable to maneuvers to increase systemic vascular resistance (knee-to-chest position), volume administration, sedation, and sometimes β-blockers. Any cyanotic spell would indicate early surgery. If the cyanotic spells are frequent or difficult to manage, then emergency surgery may be indicated. However, in neonates, often PGE2 infusion can reopen the closed PDA and stabilize the patient. In neonates who are PDA dependent from birth, surgery is indicated within a few days after birth. The small subset of patients with major aortopulmonary collaterals and multiple sources of pulmonary blood flow most often do not require surgery in the neonatal period unless they have pulmonary overcirculation or significant hypoxia.
Surgery
Initial surgical management of TOF can be either palliative or reparative. Alfred Blalock, at the persuasion of Helen Tausig, performed the first palliative systemic-to-PA shunt, widely known as the Blalock-Tausig shunt (BT shunt) to increase the pulmonary blood flow to improve the hypoxia. In this operation, the subclavian artery was transected distally, turned down, and anastomosed to the right or left PA. This historic surgery marked the emergence of cardiac surgery for congenital heart defects. Alternative shunts, such as Potts and Waterston shunts, also were advocated but fell out of favor because of complications. The modified BT shunt with the use of an expanded polytetrafluoroethylene (ePTFE) tube graft or a central aorta-to-pulmonary shunt with an ePTFE tube graft is widely practiced for palliation of not only TOF but also many other forms of congenital heart defects with decreased pulmonary blood flow.
In the current era, repair not palliation even in neonates is preferred by many large centers. Palliation with shunts carries a higher mortality than repair and has the potential for PA distortion and shunt occlusion or thrombosis, resulting in interstage morbidity or mortality.
Surgical repair consists of patch closure of VSD with an RVOT procedure. The RVOT procedure varies depending on the size of the pulmonary annulus (Figure 18-10). If the pulmonary valve annulus is better than a z score of −2 to 2.5, then pulmonary valve commisurotomy and main PA enlargement along with infundibular muscle resection are performed. This results in little or no pulmonary insufficiency and no or mild pulmonary stenosis.
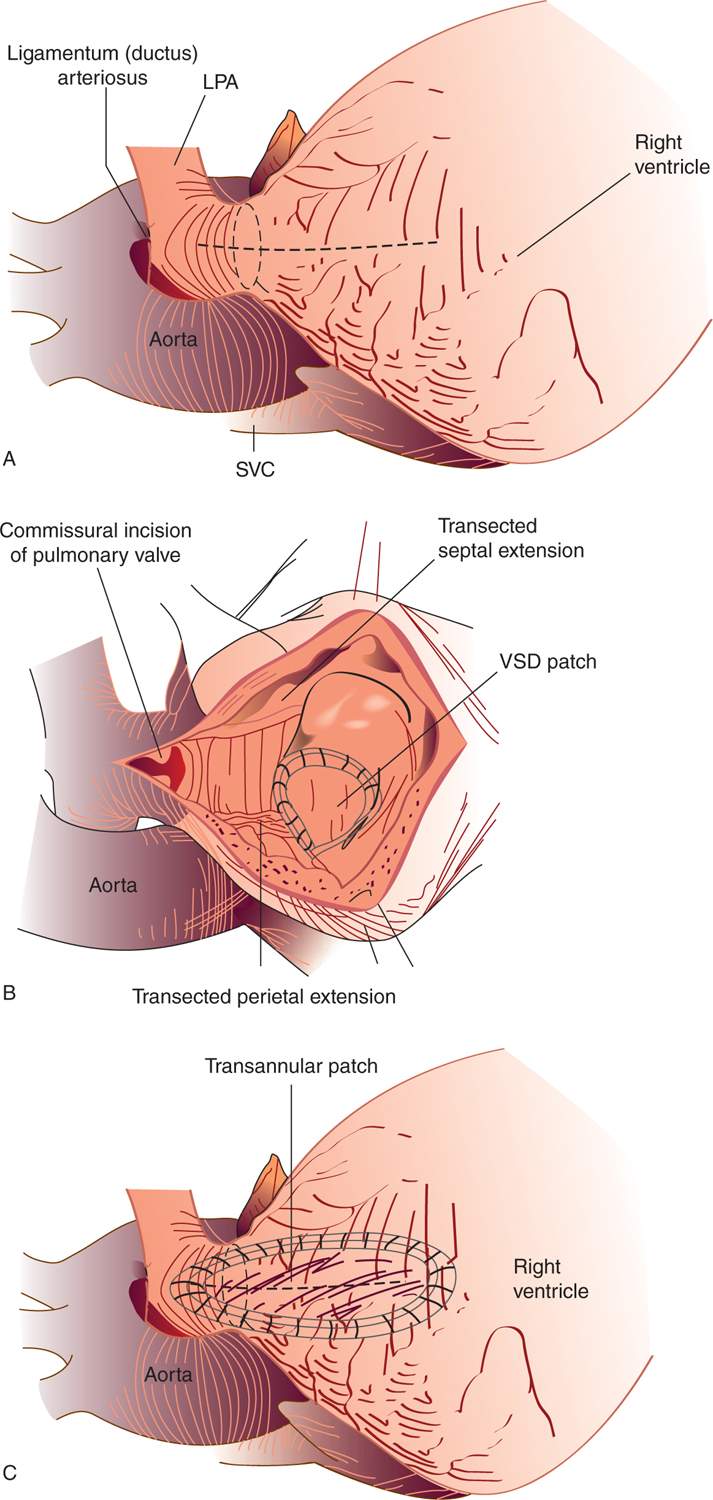
FIGURE 18-10
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


