Autoinflammatory Disorders
Philip J. Hashkes
Autoinflammatory syndromes, formerly known as periodic fever syndromes, are defined as recurrent attacks of often unprovoked systemic inflammation that are related to a lack of adequate regulation of the innate immune system.1-3 Unlike autoimmune diseases, these conditions are not generally marked by autoantibodies or autoreactive T cells. Many of these syndromes have a genetic etiology3,4 involving the excessive production and activity of interleukin (IL)-1β.5
As understanding of the pathogenesis of autoinflammatory syndromes increases, so does the spectrum of conditions that may be included within this category. Thus, systemic juvenile idiopathic arthritis, genetic inflammatory granulomatous diseases (early onset sarcoidosis and Blau syndrome) and Behçet disease (eTable 209.2  ) are now considered to be autoinflammatory disorders, although this chapter will be limited to conditions that do not fall into other diagnostic categories.
) are now considered to be autoinflammatory disorders, although this chapter will be limited to conditions that do not fall into other diagnostic categories.
These syndromes should be suspected in patients, mainly young children, with recurrent fever unexplained by infections and/or with episodic symptoms in various systems, especially the skin, gastrointestinal tract, joints, and eyes. A complete history and physical examination are crucial, and often careful determination of the organ systems involved, age of onset, length of attacks, intervals between attacks, and triggering events will allow the correct diagnosis to be suspected before genetic tests are performed (Tables 209-1 and 209-2). It is very useful to examine patients during an attack or if not possible, to ask parents to record attacks carefully in a diary and send pictures of relevant physical findings.
Autoinflammatory syndromes also should be considered in patients with unexplained elevations of acute phase reactants, even in the absence of symptoms. It is also useful to measure markers of inflammation during and between attacks, as in some syndromes attacks are only the “tip of the inflammatory iceberg” and patients consistently have increased inflammatory indices. These patients are at higher risk of developing amyloid A (AA) amyloidosis, the major adverse outcome of the autoinflammatory syndromes.
FAMILIAL MEDITERRANEAN FEVER (FMF)
Familial Mediterranean fever (FMF), first described in 1945, is the most common inherited autoinflammatory syndrome.9 It is an autosomal recessive disorder that results in recurrent attacks of fever, serositis, arthritis, and rash. An estimated 100,000 to 120,000 people have FMF worldwide. Late complications of untreated FMF include the development of renal AA amyloidosis leading to the nephrotic syndrome and renal failure. The highest prevalence of FMF is in Sephardic Jews, Armenians, Arabs, and Turks. Because of the availability of genetic diagnosis, FMF is now recognized more frequently among Ashkenazi Jews, Greeks, and Italians as well.10,11
 PATHOPHYSIOLOGY AND GENETICS
PATHOPHYSIOLOGY AND GENETICS
The familial Mediterranean fever gene (MEFV) has been localized to the short arm of chromosome 16.15-17 The product of this gene is a 781 amino acid protein termed pyrin (marenostrin in Europe). There may be a phenotype–genotype correlation with more severe disease and amyloidosis occurring in patients with the M694V, M694I, and M680I mutations.18 However, not all patients have homozygous or compound heterozygous pyrin mutations. In various series, between 30% to 70% of patients diagnosed with definitive familial Mediterranean fever by clinical criteria lack one or even both mutations, especially patients from Western Europe or the United States.19-21 In some families transmission of the condition appears to be autosomal dominant.22 Mutations or polymorphisms in genes other than pyrin may impact the development of familial Mediterranean fever or the severity of the disease, including the development of amyloidosis.18,23
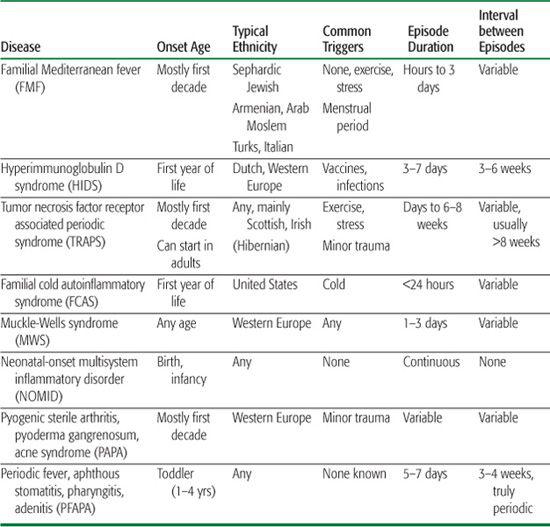
Table 209-1. Characteristics of the Main Autoinflammatory Syndromes
 CLINICAL FEATURES
CLINICAL FEATURES
Clinical signs of familial Mediterranean fever develop by age 10 in 80% of patients and by age 20 in 90%.8,27,28 A typical attack of familial Mediterranean fever results in a 12- to 72-hour attack of fever, serositis, and monoarthritis of the knee or ankle, often accompanied by an erysipelaslike rash over the involved joint. Severe abdominal pain that may mimic peritonitis or appendicitis accompanies fever in nearly 90% of patients. Other symptoms related to serositis include pleuritis (30–45% of patients), pericarditis, and scrotal swelling. Acute arthritis is seen in 50% to 75% of patients and is characterized by massive neutrophilic effusions. It usually resolves within one week, though in 5% to 10% of cases the arthritis takes a chronic course, especially in the hip and sacroiliac joints.
Since the discovery of the genetic cause of familial Mediterranean fever, additional presentations of the condition have been recognized. These include recurrent abdominal pain of childhood, recurrent arthritis without fever, prolonged febrile myalgia, and exercise-induced myalgia.7,8 Less commonly, vasculitis may be a manifestation of an MEFV mutation, especially Henoch-Schonlein purpura or polyarteritis nodosa.28-30 For unclear reasons, the course of polyarteritis nodosa related to FMF is usually more benign than the idiopathic form. Children with frequent attacks and a chronic increase of inflammatory indices often have growth abnormalities and short stature. Splenomegaly is also common.
 AMYLOIDOSIS
AMYLOIDOSIS
Prior to the discovery of colchicine as an effective therapy for familial Mediterranean fever by Goldfinger in 1972,31 amyloidosis was the major cause of morbidity and mortality. Amyloidosis usually presents with proteinuria and progresses to the nephrotic syndrome and renal failure within 3 to 5 years. Rarely familial Mediterranean fever can present with amyloidosis (“type II phenotype”). Later manifestations include signs of gastrointestinal involvement such as abdominal pain, diarrhea, malabsorption, and weight loss, as well as cardiomyopathy, macro-glossia, joint stiffness, bleeding disorders, and peripheral neuropathies.
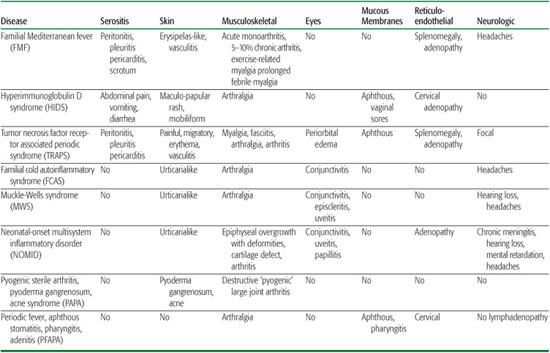
Table 209-2. System Involvement of the Main Autoinflammatory Syndromes
Risk factors for the development of amyloidosis include the particular genotype, especially patients homozygous for 694 and 680 mutations, family history of amyloidosis, geography of the patient (less amyloidosis in the United States), male gender, serum amyloid A genotype (there is an odds ratio of 7 for patients with an α/α genotype), and poor compliance with colchicine therapy.8,32
Amyloidosis should be suspected when regular screening urinanlysis demonstrates proteinuria. Renal biopsy then confirms the diagnosis, though less invasive diagnostic methods such as subcutaneous abdominal fat aspiration, rectal biopsy, and nuclear 123 I labeled scan for serum amyloid P-component may be considered.33,34 The latter method can also be used to monitor the total body load of amyloid.34 Treatment once amyloidosis has developed is generally limited to minimizing its progression by controlling inflammation. A novel agent to decrease or inhibit fibril formation by blocking polymerization of serum amyloid A, eprodisate disodium, has recently been shown to slow the decrease in creatinine clearance, though very few patients actually have a marked improvement in their amyloidosis.35
 DIAGNOSIS
DIAGNOSIS
Diagnostic criteria for familial Mediterranean fever were described in 1997 based on the clinical pattern, ethnicity, family history, and response to colchicine.36 Laboratory tests are nonspecific and reflect elevated acute phase reactants. Serum amyloid A levels may be useful in monitoring treatment efficacy.37 The diagnosis should not rely only on genetic testing because at least 30% of familial Mediterranean fever patients are either heterozygote for the MEFV gene or do not show any mutations when tested in commercial laboratories.19-21
 TREATMENT
TREATMENT
Treatment with colchicine (1–2 mg/day) is effective in preventing amyloidosis in nearly all patients and in preventing attacks in 60% to 70% of patients.31,38-42 However, 20% to 30% of patients are only partially responsive to maximal tolerated doses of colchicine, whereas 5% of patients are totally unresponsive and continue to have frequent attacks. Unresponsive patients may include those with more severe genetic mutations or patients with modifying genes that may inhibit the activity of colchicine, such as polymorphisms in MDR-1 P-glycoprotein pump transporter genes that result in lower intracellular concentration of colchicine.43 Good compliance is crucial; in some patients missing even one dose of colchicine can precipitate an attack. 
HYPERIMMUNOGLOBULINEMIA D WITH PERIODIC FEVER SYNDROME (HIDS)
Hyperimmunoglobulinemia D with periodic fever syndrome (HIDS), an autosomal recessive disease, was first described in 1984 in 6 Dutch patients.44 The mean age of onset is 6 months, with greater than 70% of patients having the first episode prior to 12 months of age.45 The disease is seen mainly in the Netherlands and Western Europe.
 PATHOPHYSIOLOGY AND GENETICS
PATHOPHYSIOLOGY AND GENETICS
HIDS is actually a metabolic disease caused by mutations in the MVK gene on the long arm of chromosome 12, encoding mevalonate kinase.46,47 The vast majority of patients have mutations in the V377I (founder gene) and I268T positions.48 Mutations result in an unstable enzyme that is less active than the wild type, particularly when patients are febrile. Hence attacks are often precipitated by infection and by vaccines that are pyrogenic.49 The cause of the fever and other symptoms is not clear, but it appears that a deficiency of geranylgeranyl and other isoprenoid substrates, rather than excessive mevalonate, is responsible for effecting inflammation.50 One theory is that this deficiency affects IL-1β processing through effects on R-type GTPase, dependent on prenylation. Total absence of MVK results in mevalonic aciduria, a syndrome characterized by severe neurologic sequelae in addition to febrile episodes (see Chapter 137).
 CLINICAL FEATURES
CLINICAL FEATURES
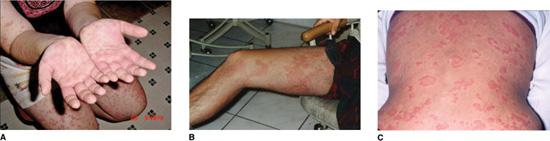
Attacks typically occur every 3 to 6 weeks and last for 3 to 7 days. Besides fever, patients develop a maculopapular, often mobiliform, rash, abdominal pain with vomiting and diarrhea, cervical lymphadenopathy, arthralgia or arthritis, and oral and genital ulceration (Fig. 209-1). Amyloidosis is a rare complication of HIDS (<3%).51
 DIAGNOSIS AND TREATMENT
DIAGNOSIS AND TREATMENT
Acute phase reactants are elevated during attacks. IgD levels are usually elevated above 100 IU/ml, and IgA is elevated in 80% to 90% of patients. Mevalonate kinase activity is decreased and urinary mevalonic acid is elevated, mainly during attacks.52 Homozygous genetic mutations are present in 75% of patients.53 However, HIDS may be seen in patients without genetic mutations when tested commercially (termed “variant HIDS”). If clinical suspicion is high, serum IgD and either MVK genetic testing or urinary mevalonic acid should be obtained.8 False-positive causes of increased IgD levels (usually not to the degree in HIDS) include other autoinflammatory diseases, HIV, diabetes mellitus, smoking, and pregnancy. In patients younger than age 3, normal IgD levels may represent a false-negative test.
NSAIDs have some symptomatic benefit. Colchicine, thalidomide, and corticosteroids are ineffective. Simvastatin may reduce the number of febrile days but has no effect on the frequency of attacks.54 Case reports also suggest occasional benefit from montelukast, etanercept, and anakinra.55,56 Regardless of treatment, attacks usually decrease in severity and frequency over time.8
TUMOR-NECROSIS FACTOR RECEPTOR ASSOCIATED PERIODIC SYNDROME (TRAPS)
First described in 1982 as familial Hibernian fever, the gene mutation for the autosomal dominant disorder tumor-necrosis factor receptor associated periodic syndrome (TRAPS) was discovered in 1999.57,58 TRAPS is the most common autosomal dominant autoinflammatory disorder. TRAPS presents in 75% of cases during the first decade of life (median age 3), but it can present at any age. There is a slight male predominance (3:2).
 PATHOPHYSIOLOGY AND GENETICS
PATHOPHYSIOLOGY AND GENETICS
The TRAPS gene (TNFRSF1A) has been localized to the short arm of chromosome 12.58 The product of this gene is the 55 kDa TNF cell membrane receptor. There is a phenotype-genotype correlation with more severe disease and amyloidosis occurring in patients with cysteine residue mutations that alter protein structure.8 Milder mutations of low penetrance, including R92Q and P46L are frequently seen in normal controls (up to 9% of the population).4 These mutations may not be pathogenic by themselves but may contribute to other still unclear causes of increased inflammation. Decreased shedding of mutated membrane-bound TNF receptors was thought to explain the unopposed serum TNF effects observed in TRAPS.59
However, it is now clear that not all mutations result in defective shedding. The pathogenesis of TRAPS may involve defects in TNF induced apoptosis and stimulation of intracellular inflammatory pathways. This also may result in a decrease in the concentration of surface receptors.
 CLINICAL FEATURES
CLINICAL FEATURES
Attacks typically last 1 to 6 weeks, and occur 2 to 6 times per year, commonly triggered by exercise.60-63 They are marked by fever as well as serositis (abdominal, chest and testicular pain), conjunctivitis, arthralgia and myalgia. Two unique features are periorbital edema and a painful, distally migrating, erythematous rash (Fig. 209-1B). Less common manifestations include recurrent pericarditis and central and focal neurologic abnormalities. Amyloidosis develops in 14% to 25% of patients, particularly those with cysteine mutations and a family history of amyloid.8
 DIAGNOSIS AND TREATMENT
DIAGNOSIS AND TREATMENT
Unlike other autoinflammatory diseases in which genetic abnormalities are only supportive of the diagnosis, the identification of TRAPS is based on finding a genetic mutation in the TNFRSF1A gene. Amyloidosis develops in 14% to 25% of patients, particularly those with cysteine mutations and a family history of amyloid.8
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


