Approach to the Child with Birth Defects
John C. Carey and Michael J. Bamshad
In the mid-1960s, David W. Smith coined the term dysmorphology to describe the field of medicine devoted to the study of abnormal human development.1 His intent was to propose a term that both replaced teratology (whose literal meaning and reference to monsters was pejorative) and captured the essence of the discipline. The purpose of this section is to summarize the principles of dysmorphology and, by doing so, provide an approach to the child with birth defects.2
Certain terms require definition in conceptualizing this approach: A syndrome is a pattern of birth defects that are etiologically related and regularly recur in different individuals (eg, Down syndrome). In other areas of medicine, the word syndrome is used less specifically and often refers to a specific set of symptoms that are not necessarily etiologically related (eg, nephrotic syndrome).
A sequence is a primary defect with a secondary cascade of structural changes. Birth defects that represent a sequence are usually localized to a single body area. Whereas a sequence can often be misinterpreted as a group of malformations, more critical inspection reveals a single malformation and a subsequent disruption or deformation. For example, the Pierre Robin sequence is caused by a primary abnormality in mandibular development that produces disruption of palatal closure and secondary obstruction of the airway by the tongue. A sequence can occur in isolation or be a component of an underlying syndrome diagnosis. For example, about 20% of children with Pierre Robin sequence have a disorder of connective tissue called Stickler syndrome (characterized by joint hyperextensibility, other skeletal manifestations and severe myopia).
An association is two or more primary defects that occur in the same individuals more often than is expected by chance. Defining a group of defects as an association suggests that the anomalies are etiologically related to one another, yet the nature and mechanism of that relationship remains unclear. For example, children with defects of the vertebrae, anus, trachea and esophagus, radius, and kidneys (renal) are often labeled with the acronym VATER association. Associations tend to be etiologically heterogenous more often than syndromes, and fewer characteristics of an association are observed in each affected child.
CLINICAL PRACTICE
The approach to a child with birth defects is multifaceted and includes the collection of phenotypic data, determination of the immediate and long-term issues of care, and the provision of the family with psychological support (Fig. 178-1). The ability to recognize and interpret minor and major anomalies is an important skill that is required for evaluating a child with a birth defect2 and is important in the collection of clinical data.
Phenotypic information that should be collected include detailed obstetrical, medical, and family histories; a comprehensive physical examination; and ancillary laboratory, physiologic, or imaging studies. The gestational and birth history needs to include exposures to over-the-counter and prescription medications as well as illicit drugs, frequency and vigor of fetal movements, intrauterine positioning, quantity and quality of amniotic fluid, maternal medical history, and the results of all prenatal testing. Documentation of at least a three-generation pedigree is also recommended. The pedigree should include information about the occurrence of sudden deaths, unexpected deaths, or early deaths (ie, deaths at younger than age 55); individuals with developmental disabilities, unusual behavioral profiles, and/or mental retardation; individuals with birth defects; degree of relatedness of parents (ie, level of consanguinity); and the ethnic background of the family. Examination of photographs of the parents taken when they were children, of siblings, and of extended family members is especially useful when attempting to determine whether a particular physical characteristic is a diagnostic clue, part of the phenotypic background of the family, or both (ie, when a parent or relative is unknowingly affected as well).
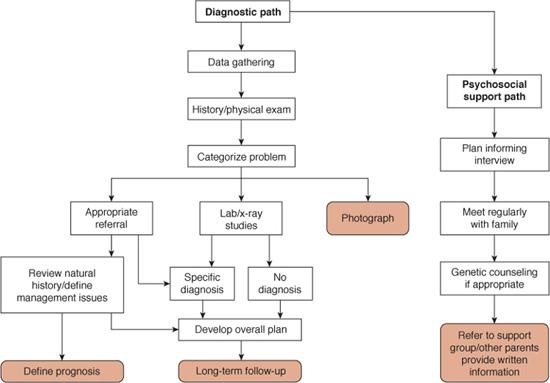
FIGURE 178-1. The approach to the management of the child with congenital anomalies. Note the diagnostic pathway and the psychosocial path are in parallel. The step involving the categorization of the problem may lead to one of the other diagnostic algorithms.
Differentiating between “normal” and “abnormal” physical findings represents the cornerstone of phenotype analysis. Many of the physical findings that are considered abnormal and clues to syndromes are found in the craniofacial region and/or limbs.3
For the cranium and face, the various relationships among the many individual components (hair, ears, forehead, nose, eyes, midface, philtral folds, lips, mouth, jaw, neck, and ears) should form a subjective impression of the overall pattern of relationships. Important questions in the clinical setting include the following: Does a particular feature (eg, a vascular dysplasia or region of marked asymmetric growth) make the face look different from what might be anticipated? Are there several features that when juxtaposed with one another make a face look particularly distinguishable? If a physical feature differs from what one considers “normal,” consider the mechanism that might have produced this impression. For example, if the ear of an infant appears set too low on the side of the head, is it because the ear is small, posteriorly rotated, or that the superior helix is overfolded? Each of these possibilities can produce the illusion that an ear is set too low. Alternatively, an ear with a normal size and structure but placed at the angle of the mandible indicates that the ear is genuinely set too low.
Each physical feature of the face (or hands or chest, etc) can be classified as to whether the variation is a minor anomaly or mild malformation. Minor anomalies include both qualitative characteristics (eg, size of the nasal tip) that can be challenging to judge as normal versus abnormal as well as features that lend themselves to rapid quantitative measurement (eg, distance between the inner canthal folds).3,4 Measurement of many of the quantitative features can be compared with normative data collected from selected groups of individuals considered normal.1,3,5 The handbook by Hall et al. provides a particularly valuable resource that summarizes the graphs and guidelines for measurements needed in the clinical setting.5 Each physical characteristic must be considered in the context of the general morphology of the family, their ethnic group, and their continent of origin. Many of the physical findings that are used to distinguish among persons of recent Asian or African ancestry are quantitative traits that may differ substantially between groups, alone or in combination. For example, short palpebral fissures are a consistent finding in children with fetal alcohol syndrome. However, wide variation exists in the length of the palpebral fissures among individuals of European, Asian, and/or Native American ancestry, which makes the interpretation of the importance of short palpebral fissures challenging.3 In contrast to minor anomalies, mild malformations are structural changes of a mild degree; these findings have no intrinsic medical significance (unlike major malformations). Examples include auricular tags and posterior polydactyly.4
The measurement of a body part or the relationships between parts is an important component of accurate diagnosis.3 For example, the length of the ear can be a valuable clue when evaluating a child for fragile X syndrome (characterized by a long ear) or trisomy 21 (characterized by a short ear). Apparently wide-spaced eyes can relate to the presence of epicanthal folds, telecanthus (soft-tissue displacement of the inner canthi), short palpebral fissures, or true ocular hypertelorism. Distinction among these possibilities requires astute observation, knowledge of normal facial structure relationships/proportions, and measurements of the inner canthal/interpupillary and outer canthal distances. These distinctions are crucial, because different syndromes are characterized by different abnormalities of eye placement.
Examination of the limbs should be conducted similarly. The general relationships among the parts of the hand (eg, length of digits, spacing between digits) should be assessed and subsequently each part should be examined individually.6 The thumb is a particularly complex digit that requires skill in differentiating between normal variations and abnormal findings. Alterations of the thumb include low-set thumbs (usually hypoplastic with an underdeveloped thenar eminence), tapered thumbs, bifid thumbs, and triphalangeal thumbs (three phalanges instead of two). Examination of the flexion creases on the digits including the thumbs is also a valuable observation because these creases develop between 8 and 10 weeks of embryogenesis, and perturbations of the flexion creases (eg, hypoplasia, absence) reflect an abnormality of fetal movement in the first trimester of gestation. For example, children with multiple congenital contractures caused by a reduced quantity of amniotic fluid in the third trimester may have normal flexion creases, whereas an infant with amyoplasia (a form of arthrogryposis) may have complete absence of the flexion creases.
The recognition of minor and major anomalies helps to determine whether a child may have a multiple congenital anomaly syndrome and, if so, how to proceed toward confirmation of a specific diagnosis. Comparing physical findings in members of a child’s extended family may indicate that a child who apparently has multiple anomalies simply has an apparently abnormal appearance related to combined effects of the parents’ atypical but normal physical characteristics. This underscores the point that overinterpretation of multiple minor anomalies in a child is troublesome. Although 10% of newborns have two or more minor anomalies, the overwhelming majority of these otherwise normal-appearing infants do not have a specific syndrome and are unlikely to experience further physical and/or developmental problems.
Minor anomalies often provide useful clues as to whether a more severe defect of a body part or organ may be present. Many purported associations have been overstated in the past and probably have little significance (eg, the purported association of accessory nipples and renal defects). However, some minor anomalies are clearly markers for more important underlying defects.4 For example, the presence of a deep sacral dimple, commonly with an overlying patch of hair or a vascular stain, should prompt further investigation for spinal dysraphism. Pits in the skin or tags of skin near the external ear should bring to mind specific syndrome diagnoses (eg, branchial-oto-renal syndrome) and are associated with an increased risk of hearing loss.
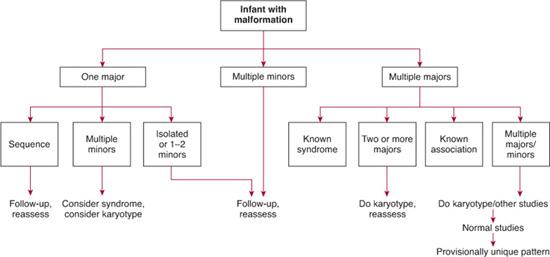
The most pivotal step in the diagnostic algorithm of a child with multiple congenital anomalies is categorizing the constellation of findings into an organized pattern of malformation (whether characteristic of a recognizable syndrome of known etiology or not) (Figs. 178-1 and 178-2). Concluding that a child’s phenotypic findings probably fit a pattern of multiple anomalies in contrast to representing an isolated defect is critical (Fig. 178-2). A child with an isolated finding (eg, isolated cleft lip) may require no further diagnostic evaluation. In contrast, a child with multiple phenotypic findings consistent with an organized pattern of presumably etiologically related defects may need substantially more testing to reach a diagnosis. For example, diagnostic testing, that is, high-resolution chromosomes or comparative genomic hybridization-CGH-microarray to look for aneuploidy, deletions, or duplications, is virtually always indicated in children with an un-diagnosed pattern of multiple congenital defects. A skeletal survey may help resolve the diagnosis in a child with a limb abnormality or who is suspected of having a skeletal dysplasia. Various online resources are available for helping in the diagnostic process.7,8
Most children who are being evaluated for multiple congenital anomalies do not need to be tested for an inborn error of metabolism (see Chapter 134). For example, children who have disorders of amino acid metabolism do not present typically with birth defects. However, some conditions resulting from inborn errors of intermediary metabolism do present with malformations and/or dysplasias (see Chapter 134 and Table 176.1). To complicate matters further, some very well-known multiple congenital anomaly syndromes have turned out to be caused by defects of metabolism. For example, Smith-Lemli-Opitz (SLO) syndrome, characterized by multiple malformations and variable cognitive abnormalities, is caused by a defect of cholesterol biosynthesis resulting from mutations in the gene that encodes 7-dehydrocholesterol reductase.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


