Antibacterial Therapy
David K. Hong and Charles G. Prober
The first antibiotic to be discovered was penicillin, a natural product of Penicillium mold. Innumerable microbial products have been investigated since then, and much work has been done in chemically modifying these natural products in an attempt to enhance the benefits while minimizing the undesirable effects. These modified products, termed semisynthetic antibiotics, increased stability and solubility, improved pharmacokinetics (ie, wider distribution and longer half-life), and increased antimicrobial activity. Minimizing the undesirable effects creates antibiotics with decreased toxicity and increased efficacy.
Unfortunately, overuse of this vast array of antibiotics now is one of our most pressing problems. Antibiotics are the most commonly prescribed class of drugs in the United States. In children, antibiotics represent about 30% of all prescribed drugs. Almost half of children under age 15 will have an antibiotic prescribed to them each year.1
Misuse of antibiotics is common. Thirty percent to 65% of antibiotic prescriptions in hospitals are found to be irrational, inappropriate, or of questionable value. In community practice, market research data have determined that 50% of physicians prescribe antibiotics for the common cold, although this trend has improved in recent years.1,2 The reasons for this antibiotic “abuse” are multifactorial, but the desire to help patients, fear of missing a bacterial infection that might respond to antibiotics, and the ease of treating a possible bacterial infection versus considering and investigating an alternative diagnosis all contribute.
One prevalent attitude is that the risk of not treating an infection is greater than the risk of side effects from antibiotic treatment. In fact, approximately 5% of patients taking antibiotics experience side effects, and the indiscriminate use of antibiotics alters the drug-resistance patterns of isolates from the individual being treated and from the environment in general. Furthermore, a potentially more serious infection such as meningitis can be masked by incidental antibiotic therapy.
GENERAL PRINCIPLES OF ANTIBIOTIC THERAPY
 ANTIBIOTIC SELECTION
ANTIBIOTIC SELECTION
The decision to prescribe an antibiotic is based on proof or strong suspicion that the patient has a bacterial infection. Probable viral infectious or noninfectious processes should not be treated with antibiotics. However, in the critically ill patient in whom there is some chance that a bacterial infection may be a contributing factor, it is prudent to administer antibiotics effective against the most likely pathogens.3
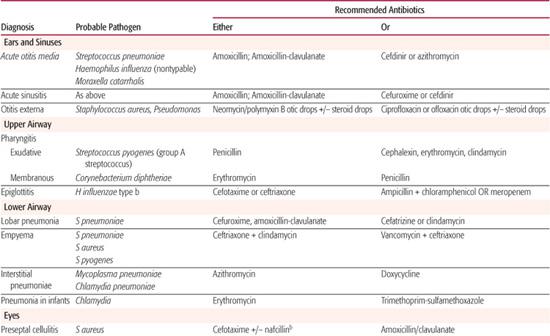
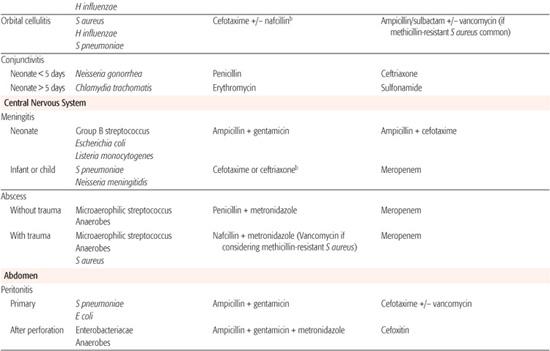
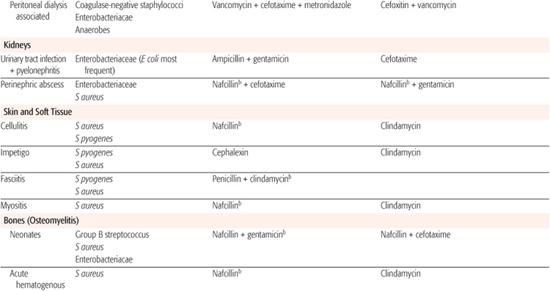
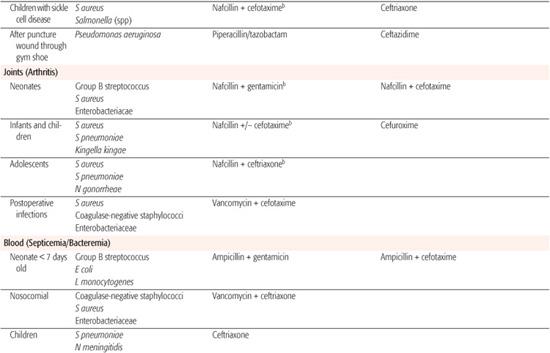
Whenever possible, antibiotic selection should be based on the isolation of a pathogen, but most patients who require antibiotic therapy present with an acute problem that mandates initial empiric therapy. The antibiotic(s) chosen should be based on the pathogens likely to be responsible for the infection, knowledge of local antibiotic sensitivity patterns, and specific host characteristics. If more than one antibiotic is active against the likely pathogens at the site of infection, the specific agent should be chosen on the basis of relative toxicity, convenience of administration, and cost. Once the pathogen is identified, the antibiotic with the most narrow spectrum of activity should be used. Table 246-1 outlines one set of suggested drugs of choice for a wide variety of childhood infections.4
 ROUTE OF ADMINISTRATION
ROUTE OF ADMINISTRATION
The route chosen for the administration of antibiotics depends on a number of factors, including the severity of infection, drug levels required for therapy, logistics of administration, and anticipated patient compliance. Outpatient therapy usually is given orally except when a single intramuscular injection may suffice. Long-term administration of intravenous antibiotics also has been accomplished in the outpatient setting. In the sick hospitalized patient, the intravenous route is commonly used because it ensures direct delivery of the antibiotic, and, in general, the blood concentration of antibiotic attained is higher. Patients who do not have established intravenous access can have antibiotics administered intramuscularly, unless they have a bleeding disorder or are in shock. If treatment is likely to be prolonged, frequent intramuscular injections are uncomfortable for the patient, and the intravenous route is preferable.
Increasingly, antibiotics are initially given by the parenteral route until the patient is stable. The oral route is then used to complete the course of therapy. This treatment protocol has been used most frequently in the therapy of osteomyelitis and septic arthritis. Compliance must be assured, the adequacy of antibiotic absorption must be assessed frequently, and the patient must have frequent clinical examinations. The advantages are self-evident: technical demands related to prolonged maintenance of an intravenous access route are reduced, as are risks of thrombophlebitis, catheter-associated infections, and the duration of hospitalization.
 DURATION OF THERAPY
DURATION OF THERAPY
The duration of antibiotic administration recommended for specific infections is often based on uncontrolled experience, not on controlled trials. Guidelines concerning the duration of therapy for most infections are outlined in this text. However, clinicians should not commit patients to a rigid duration of therapy when the infection is first diagnosed; therapy should be guided by clinical response rather than by an arbitrary number of days.
Clinical monitoring usually involves sequential physical examinations with special reference to the site originally infected and body temperature. Signs of inflammation and fever should resolve within several days after appropriate antibiotics are initiated. Laboratory monitoring may include repeat bacterial cultures to ensure sterilization, and for severe infections, it may be useful to monitor the peripheral white blood cell count and acute-phase reactants (eg, erythrocyte sedimentation rate [ESR] or C-reactive protein [CRP]). A lack of response to therapy may mandate a change of antibiotics.
CLASSIFICATION OF ANTIBIOTICS
Antibiotics target unique bacterial synthetic processes that differ from those in human cells. This directed attack is referred to as selective toxicity. The four most common sites of antibacterial action that have been targeted for antibiotic development are inhibition of the synthesis of the bacterial cell wall, nucleic acid, protein, and folate (Table 246-2). Antibiotics can be classified by mechanism of action or as either bacteriostatic or bactericidal. Bacterio-static agents inhibit bacterial cell replication but require the host’s immune factors to clear the infection, whereas bactericidal agents kill the bacteria. If host immunity is suppressed or the infection is in an area of poor immunologic surveillance (eg, cerebrospinal fluid), bacteriostatic agents may not be as effective as bactericidal agents.
Table 246–1. Drugs of Choice: A Prescribing Guide

Chloramphenicol and erythromycin are bacteriostatic against most bacteria, although chloramphenicol is bactericidal against Haemophilus influenzae type b, Streptococcus pneumoniae, and Neisseria meningitidis. Bactericidal antibiotics include penicillins, cephalosporins, vancomycin, and aminoglycosides. They cause microbial death by cell lysis. Some antibiotics, such as the sulfonamides and tetracyclines, may be either bacteriostatic or bactericidal, depending on the concentration of drug, the nature of the environment, and the specific bacteria against which they are being used.
ANTIBIOTIC RESISTANCE
The development of microbial drug resistance results from the widespread use of the growing array of antimicrobial agents, coupled with the ability of bacteria to acquire and spread resistance in addition to the capacity of humans to spread bacteria. Antimicrobial drug resistance represents the greatest threat to successful antibiotic therapy and is a major driving force behind the search for newer drugs. With few exceptions, such as the continued activity of penicillin against group A streptococci, most bacteria have developed resistance to previous agents of first choice. One of the most recent and dramatic examples of this seemingly inevitable development of resistance is the emergence and increasing prevalence of community-associated methicillin-resistant Staphylococcus aureus (MRSA).5 The potential consequences of antibiotic resistance for the individual patient are an increased likelihood of hospitalization, a longer hospital stay, and about a twofold increased death rate. Furthermore, the treatment of drug-resistant bacteria often demands the use of more toxic and expensive drugs.
Developing countries and hospitals have become common breeding grounds and reservoirs for antimicrobial-resistant pathogens. Clinical isolates commonly are resistant to a number of antimicrobial agents. “Multiresistance” usually arises when the same mechanism confers resistance to several agents or when individual resistance genes cluster on either the bacterial chromosome or on extrachromosomal resistance plasmids (R-plasmids).
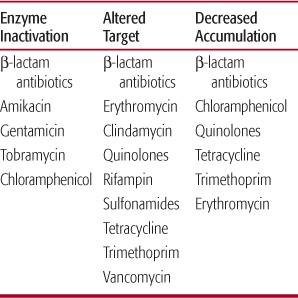
There are a limited number of mechanisms by which bacteria develop resistance to antibiotics. In very general terms, these mechanisms include (1) the production of enzymes that inactivate or modify the antibiotic, (2) decreased antibiotic uptake or an active efflux system, and (3) alteration in antibiotic target (Table 246-3).6 β-Lactamases probably are the best known inactivating enzymes produced by resistant bacteria. Bacterial resistance to penicillins and cephalosporins is often mediated by these enzymes. Alterations in outer-membrane proteins can decrease penetration of antibiotics into the bacteria. An example of an alteration of the antibiotic target may also result in resistance. Strains of penicillin-resistant S pneumoniae have a markedly reduced affinity for the penicillin-binding protein. Bacteria may develop resistance mediated by more than one mechanism.
USE OF ANTIBIOTICS IN COMBINATION
Usually a single antibiotic can be prescribed to treat an uncomplicated infection caused by a single pathogen. The most common reason for combining two or more antibiotics is to assure adequate therapy until the infecting pathogen has been identified. Combination therapy also may be necessary when the infection is presumed or proved to be caused by more than one bacterium that cannot be adequately treated with a single agent. Pelvic and intra-abdominal infections, usually caused by a mixture of aerobes and anaerobes, are examples. Combining two agents theoretically may prevent or delay the emergence of resistance, justifying the use of two drugs to treat Pseudomonas aeruginosa or mycobacterial infections. Antibiotics also are prescribed in combination in the hope that there will be greater inhibition or killing of the pathogenic bacteria than would occur with single-drug therapy. An example is treatment of enterococcal infections with a penicillin plus an aminoglycoside. Disadvantages of combination antibiotic therapy include an increased incidence of superinfection and toxicity, increased cost, and potential adverse drug interactions.
GUIDANCE IN ANTIBIOTIC USE
The rational use of antibiotics requires knowledge of their spectrum of activity, certain aspects of their pharmacokinetics, their most common side effects, and their cost as compared to agents of equal safety and efficacy. Another consideration is the pharmacodynamic characteristics of a particular antibiotic, with the goal being to optimize exposure of the drug over time to maximize therapeutic efficacy. In children, appropriate oral formulations are particularly relevant. The seasoned clinician will depend on a small number of antibiotics that have established reliability. The newest antibiotics are not necessarily the best, although they are often among the most expensive.
Table 246–2. Classification of Antibiotics by Mechanism of Action
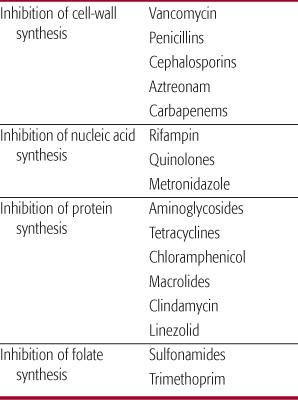
Table 246–3. Mechanisms of Resistance to Antimicrobial Agents
The minimum kinetic knowledge required for frequently prescribed antibiotics includes the following:
• The expected concentration of the antibiotic at the site of infection that will be attained after the selected dose. This implies knowledge of the serum concentrations attained and the diffusion characteristics of the antibiotic into infected tissue. The adequacy of the anticipated concentration of drug at the site of infection is determined by the antibiotic sensitivity pattern of the infecting bacterium.
• The half-life (t1/2) of the antibiotic (see Table 246-5). In general, antibiotics should be administered every third half-life.
• The sources of pharmacokinetic variation, knowledge of which necessitates some understanding of excretion and metabolism. If an agent is excreted primarily by the kidneys or by the liver, and the patient has compromised renal or hepatic function, the dose may have to be adjusted. Some host variables that influence the kinetics of different antibiotics are outlined in Table 246-4. The use of antibiotics in infants whose organ maturity is evolving presents a special challenge to clinicians.
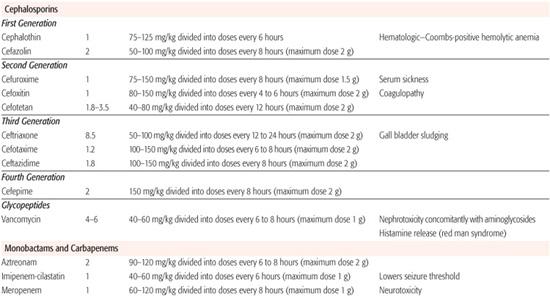
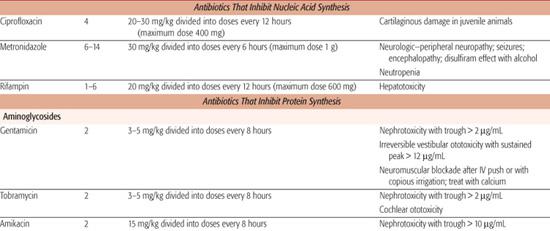
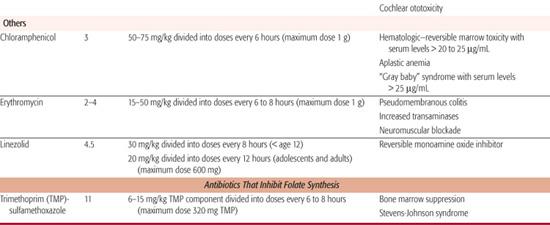
Many factors determine the correct dosage of antimicrobials, including age (dosages for premature and newborn infants differ from those for older children), weight, route of administration, and pharmacodynamic characteristics of the agent. Significant liver or renal disease often requires adjustments in dose. Parenteral dosages of several commonly used antimicrobials are listed in Table 246-5.
SPECIFIC ANTIBIOTICS
 PENICILLINS
PENICILLINS
Penicillin G is the “natural” or “native” penicillin; all other penicillins are semisynthetic compounds. The basic structure of penicillin consists of a 6-aminopenicillanic acid (6-APA) nucleus and a variety of side chains. The 6-APA nucleus has a thiazolidine ring connected to a β-lactam ring. The integrity of the β-lactam ring is necessary for antibacterial activity. Hence organisms that produce β-lactamases, which break the ring configuration, render the drug inactive.
The penicillins are divided into three groups on the basis of their antibacterial spectrum as detailed in the following discussion.
Narrow-Spectrum, β-Lactamase-Sensitive Penicillins
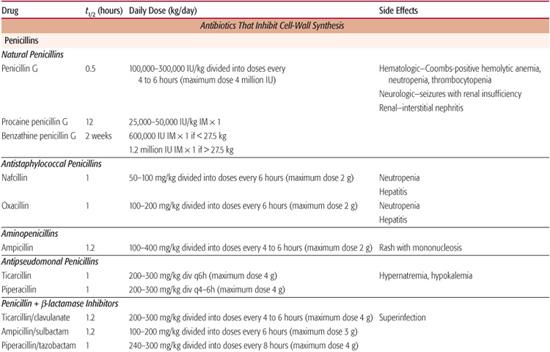
The prototype of this group is penicillin G. This antibiotic is active against most Gram-positive bacteria with the exception of penicillinase-producing S aureus. In recent years, an increasing proportion of isolates of S pneumoniae have developed relative or absolute resistance to penicillin. Penicillin G is also active against most Neisseria species, some Gram-negative anaerobes, and Treponema pallidum. Penicillin G is not active against most Gram-negative aerobic organisms. Bacteria sensitive to penicillin generally have a minimal inhibitory concentration (MIC) less than 0.05 mg/L.
A 100,000-IU/kg dose of penicillin G (1 IU = 0.6 μg) administered intravenously results in serum concentrations in excess of 10 mg/L, 200-fold higher than the minimal inhibitory concentration of most sensitive bacteria. This antibiotic also diffuses widely, attaining therapeutic concentrations in most body tissues. For example, up to 25% of serum concentrations are attained in the cerebrospinal fluid during the treatment of bacterial meningitis. The t1/2 of penicillin G is less than 1 hour, and it is eliminated primarily by renal tubular secretion. This secretion can be inhibited by probenecid. Because renal dysfunction will compromise the elimination of penicillin, dosages may need to be reduced in patients with renal insufficiency. This is necessary only in the most extreme circumstances, owing to the low toxicity of penicillin.
Penicillin V, the phenoxymethyl analog of penicillin G, is much more stable than is its parent compound and therefore better absorbed from the gastrointestinal tract. A 250-mg dose of this preparation results in concentrations roughly equivalent to those attained after two doses of orally administered penicillin G. Procaine penicillin is a commonly used intramuscular preparation that produces low (3 mg/L) concentrations of drug sustained over several days. It is best suited to the single-dose outpatient treatment of very sensitive organisms (eg, group A streptococci). Benzathine penicillin is another preparation given intramuscularly. Serum concentrations of less than 0.1 mg/L, sustained for as long as 3 to 4 weeks, are attained with this formulation. It is used to prevent recurrent group A streptococcal infections in patients with rheumatic fever.
The most frequent indications for the use of penicillin G and its derivatives in children are for infections caused by most species of streptococci and infections caused by sensitive Neisseria species. However, in geographic areas where the incidence of penicillin-resistant Neisseria gonorrhoeae exceeds 10%, empiric therapy with penicillin is not recommended.
Table 246–4. Some Variables That Influence the Kinetics of Antibiotics
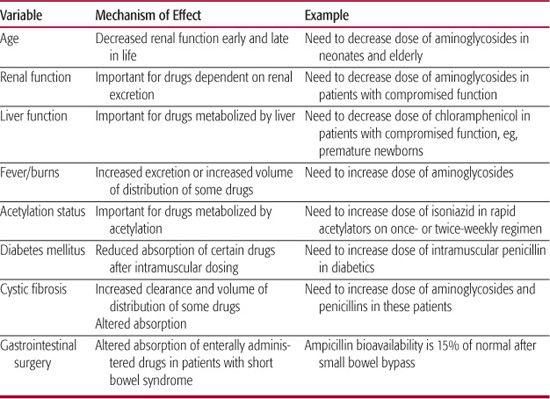
Table 246–5. Parenteral Doses of Antibiotics
Broad-Spectrum, β-Lactamase-Sensitive Penicillins
Aminopenicillins
Examples of aminopenicillins include ampicillin and amoxicillin. The activity of the aminopenicillins against Gram-positive bacteria is similar to that of penicillin. Aminopenicillins are, however, more active against enterococci, Listeria monocytogenes, and non-β-lactamase-producing H influenzae. They also are active against some Escherichia coli, Shigella, Salmonella, and indole-negative Proteus species. The minimal inhibitory concentrations necessary against Gram-negative organisms are usually in the range of 1 to 5 mg/L.
The serum concentration of ampicillin after a 1-g intravenous dose is approximately 40 mg/L; after a 500-mg dose taken orally, it is approximately 4 mg/L. Concentrations of amoxicillin are usually twice those of ampicillin after an equivalent oral dose. The distribution, t½, and excretion characteristics of the aminopenicillins are similar to those of penicillin.
Ampicillin and its derivatives are among the most useful antibiotics for treating children with infections caused by sensitive Gram-negative aerobic bacteria, enterococci, L monocytogenes, and β-lactamase-negative H influenzae. Amoxicillin is the drug of choice for the treatment of acute otitis media.
Carbopenicillins and Ureidopenicillins
Carboxypenicillins are represented by carbenicillin and ticarcillin; ureidopenicillins are represented by piperacillin, azlocillin, and mezlocillin. These antibiotics have a broader spectrum of Gram-negative activity than do the aminopenicillins, and include activity against most strains of P aeruginosa. The usual minimal inhibitory concentrations of P aeruginosa range from 12 to 25 mg/L, with piperacillin consistently being the most active agent. Maximum serum concentrations of these antibiotics are usually in excess of 150 mg/L, after a dose of 3 to 5 g. These antibiotics are used almost exclusively in the treatment of urinary tract, lung, and bloodstream infections caused by ampicillin-resistant enteric Gram-negative pathogens.
β-Lactamase-Resistant Penicillins
These penicillins include nafcillin, oxacillin, methicillin, cloxacillin, dicloxacillin, and flucloxacillin. The principal bacteriologic advantage of this group of antibiotics is their activity against β-lactamase-producing staphylococci. Most isolates of methicillin-sensitive S aureus have minimal inhibitory concentrations of 0.25 to 0.5 mg/L. These antibiotics are less active than penicillin G against the other Gram-positive bacteria, and they are inactive against Gram-negative enteric organisms. Maximum serum concentrations after a 1-g intravenous dose of nafcillin, methicillin, or oxacillin range from 20 to 40 mg/L; whereas after a 500-mg oral dose of cloxacillin or oxacillin, they range from 4 to 8 mg/L. Dicloxacillin and flucloxacillin have an enhanced absorption after oral administration. Serum concentrations of these agents are twice those of cloxacillin or oxacillin after an equivalent oral dose. These penicillins are used almost exclusively for the treatment of mild, moderate, and severe infections caused by methicillin-sensitive S aureus, including cellulitis, osteomyelitis, pneumonia, and septicemia.
Toxicity of Penicillins
The adverse reactions of all penicillins are similar. In general, these agents are well tolerated; however, suspension formulations tend to have an unpleasant taste and aftertaste and, as a result, may be poorly accepted. All penicillins have a wide toxic-to-therapeutic ratio, although they can cause hypersensitivity reactions, neurotoxicity, nephrotoxicity, and hematologic toxicity.
Hypersensitivity reactions are relatively common and include rashes, serum sickness, anaphylaxis, nephritis, and drug fever. Urticarial skin reactions and anaphylaxis, which occur within 20 to 30 minutes after a dose, are termed immediate reactions. These are the most dangerous reactions and constitute absolute contraindications to future treatment with a penicillin derivative. Fortunately, the incidence of anaphylaxis is only 0.01% to 0.02% of individual courses of therapy.7
Nonurticarial skin eruptions that occur several days after the initiation of a course of penicillin are relatively common and do not preclude future therapy with penicillins. Many such eruptions represent the rash of a viral infection for which an antibiotic has been inappropriately prescribed. Patients manifesting these sorts of reactions should not be labeled “penicillin-allergic.”
Convulsions and other forms of central nervous system irritation may occur when high doses of a penicillin have been administered, particularly to patients with compromised renal function. Reactions are also more likely when high cerebrospinal fluid concentrations of drug are attained, such as in patients with meningeal inflammation.
Interstitial nephritis can occur during the course of therapy with any penicillin, although it is most frequently associated with the administration of methicillin. Hypokalemia is another renal side effect of high-dose penicillin therapy that results from penicillins acting as nonresorbable anions.
Coombs-positive hemolytic anemia may occur with any of the penicillins, as may neutropenia. Neutropenia is most common among patients receiving a β-lactamase-resistant penicillin and usually resolves when the antibiotic is stopped. Decreased platelet aggregation, which may precipitate bleeding, has been noted at high concentrations of most penicillins. It is most marked with carbenicillin and ticarcillin.
In addition to the reactions noted above, which are common to all of the penicillins, ampicillin or amoxicillin can cause a characteristic nonurticarial maculopapular rash that does not appear to have an allergic etiology. This rash usually appears 3 to 4 days after the onset of therapy and is more frequent in patients with viral infections, especially infectious mononucleosis.
 CEPHALOSPORINS
CEPHALOSPORINS
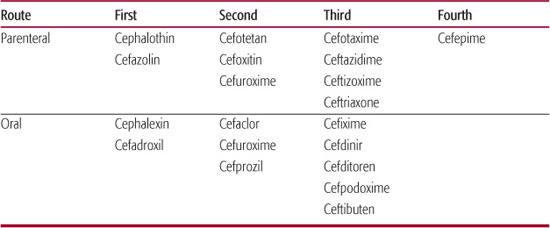
The cephalosporins are currently divided into four generations, with original agents being referred to as first-generation cephalosporins, and the most recent agents as fourth-generation (see Table 246-6). In general, the spectrum of activity of the cephalosporins increases with each generation because of decreasing susceptibility to bacterial β-lactamases.
First-Generation Cephalosporins
These cephalosporins are active against most staphylococci and pneumococci and all streptococci, with the important exception of enterococci. Minimal inhibitory concentrations against sensitive Gram-positive organisms are usually less than 0.5 mg/L. Their activity against aerobic Gram-negative bacteria and against anaerobes is limited. Maximum serum concentrations after a 500-mg dose of oral cephalexin are approximately 20 mg/L, whereas they are 100 mg/L after 1-g intravenous doses of cefazolin. These antibiotics distribute widely throughout the body but do not penetrate well into the cerebrospinal fluid. Therefore, they must not be used to treat meningitis. Their t1/2 ranges from 30 minutes to 1.5 hours, and they are eliminated unchanged in the urine. Doses may need adjustment in the presence of renal insufficiency, although these agents have a wide toxic-to-therapeutic ratio.
The first-generation cephalosporins are rarely drugs of first choice. They may, however, be useful in patients who are intolerant to penicillins. Although cephalosporins and penicillins share the β-lactam ring structure, the true incidence of cross-reactivity to cephalosporins in skin-test-confirmed penicillin allergic patients is roughly 4%. Cephalosporins should not be administered to patients with a history of IgE-mediated hypersensitivity reactions to penicillins, as similar reactions to cephalosporins may be observed. These antibiotics are useful in the perioperative prophylaxis of surgical procedures that carry a high risk of postoperative infections caused by staphylococcal species, such as those involving the cardiovascular system and bones.
Second-Generation Cephalosporins
These cephalosporins have a broader bacterio-logic spectrum than do the first-generation agents. For example, cefuroxime and cefaclor not only are more active against Gram-negative enteric bacteria but are active against both β-lactamase-negative and -positive strains of H influenzae, generally at concentrations below 2 mg/L. The major bacteriologic advantage of the cephamycins, cefoxitin and cefotetan, is their activity against a broad range of anaerobic pathogens, most anaerobes being inhibited by less than 16 mg/L. Maximum serum concentrations of cefuroxime and cefoxitin after a 1-g intravenous dose are approximately 100 mg/L. Concentrations of cefaclor are approximately 10 mg/L after a 200-mg oral dose. The half-lives of the second-generation agents are similar to those of the first-generation agents. Excretion of second-generation cephalosporins is primarily renal, and they distribute widely. However, they do not attain sufficient concentrations in the cerebrospinal fluid to warrant their use in the treatment of bacterial meningitis.
Table 246–6. Representative Cephalosporins Classified by Generation
Second-generation cephalosporins, like the first-generation agents, are rarely drugs of first choice. Cefuroxime, because of its activity against Gram-positive cocci and H influenzae, has been actively promoted as a good agent for the treatment of a variety of infections in children, including cellulitis, osteomyelitis, septic arthritis, and pneumonia. However, it is not recommended for the therapy of bacterial meningitis, because of several reports of bacteriologic failures. The most common use of cefaclor is in acute otitis media. However, other, less expensive, better tolerated, and equally efficacious agents are available. Cefoxitin and cefotetan are effective agents in the prevention and treatment of intra-abdominal or pelvic infections.
Third-Generation Cephalosporins
Third-generation cephalosporins retain much of the Gram-positive activity of the first two generations, although their antistaphylococcal activity is reduced 5- to 10-fold. They are remarkably active against most Gram-negative enteric isolates, with minimal inhibitory concentrations usually less than 0.5 mg/L. Some third-generation cephalosporins (eg, ceftazidime) also are active against most isolates of P aeruginosa. Maximum serum concentrations of the third-generation agents range from 50 to 150 mg/L after a 1-g intravenous dose. In healthy subjects, their half-lives range from 1 hour (cefotaxime) to between 6 and 8 hours (ceftriaxone). These antibiotics diffuse well into most tissues, in contrast to members of the first two generations. Cefotaxime and ceftriaxone, in particular, penetrate well into the cerebrospinal fluid. Excretion of these agents is primarily renal.
The possible indications for third-generation cephalosporins include empiric therapy of suspected bacterial meningitis, treatment of hospital-acquired multiple-resistant Gram-negative aerobic infections, and suspected infections in certain compromised hosts (eg, those with fever and neutropenia). Ceftriaxone also is the drug of choice in treating infections caused by N gonorrhoeae in geographic areas with a high incidence of penicillin-resistant isolates.
Although these antibiotics show excellent activity against a wide variety of enterobacteriaceae, their widespread use has led to the development of antibiotic resistance. These plasmid-encoded extended-spectrum β-lactamases (ESBLs) are considered resistant to all cephalosporins and have rapidly spread over the past decades, especially among strains of E coli and Klebsiella pneumoniae. Although their prevalence has been low in US pediatric populations, clinicians should continue to be judicious in their use of third-generation cephalosporins. All microbiology laboratories should screen for the presence of ESBL-producing enterobacteriacae.
Fourth-Generation Cephalosporins
This newest generation of cephalosporins combines the antistaphylococcal activity of first-generation agents with the Gram-negative spectrum (including Pseudomonas) of third-generation cephalosporins. Currently cefepime is the only available fourth-generation cephalosporin. Cefepime has excellent activity against multiple-resistant Gram-negative bacteria including P aeruginosa, and bacteria that produce the AmpC-inducible β-lactamase. Possible indications for use include the therapy of infections suspected or proved to be caused by multiple-resistant pathogens or in treating suspected infections in immunocompromised hosts at risk of infections caused by Pseudomonas species.
Complications of Cephalosporins
Serious adverse reactions to the cephalosporins are uncommon. As with most antibiotics, the full spectrum of hypersensitivity reactions may occur, including rashes, fever, eosinophilia, serum sickness, and anaphylaxis. Allergic reactions are seen in approximately 5% of courses. Adverse reactions attributable to irritation at the site of administration are common. These reactions include local pain after intramuscular injection, phlebitis after intravenous administration, and minor gastrointestinal complaints after oral administration.
Therapy with cephalosporins leads to the development of a positive direct Coombs reaction during approximately 3% of courses. This is, however, not commonly associated with hemolytic anemia. Some of the cephalosporins are associated with dose-related nephrotoxicity, whereas others are associated with an interstitial nephritis.
The third-generation drugs may cause transient elevations of liver function test results and blood urea nitrogen concentrations. Ceftriaxone has been associated with gallbladder sludging. These broad-spectrum cephalosporins also have a profound inhibitory effect on the vitamin K–synthesizing bacterial flora of the gastrointestinal tract.
 β-LACTAMASE INHIBITORS
β-LACTAMASE INHIBITORS
β-Lactamase inhibitors competively inhibit β-lactamase enzymes, restoring the original spectrum of activity to enzyme-susceptible antibiotics.
Currently marketed inhibitors include clavulanic acid in fixed combination with either amoxicillin or ticarcillin, and sulbactam in fixed combination with ampicillin. Piperacillin is also available in combination with the β-lactamase inhibitor tazobactam. Although amoxicillin-clavulanate is effective in the therapy of such infections as otitis media, sinusitis, lower respiratory tract infections, and skin and soft-tissue infections, equally effective and less costly alternatives for these infections are generally available. However, some infections are polymicrobial and may involve anaerobes; for these the addition of a β-lactamase inhibitor may be of value. These infections include infected animal and human bites, odontogenic infections, chronic sinusitis, and intra-abdominal infections.
The side effects of these agents reflect those of their parent compounds. Gastrointestinal disturbances, especially diarrhea, are common among those receiving orally administered β-lactamase inhibitors. It appears that these symptoms can be partially ameliorated by giving the drug with food and following each dose with 2 to 4 ounces of fluid.
 VANCOMYCIN
VANCOMYCIN
The primary activity of this cell-wall-active antibiotic is against Gram-positive bacteria. Most clinical isolates of S aureus and coagulase-negative staphylococci are inhibited by less than 1.6 mg/L of this antibiotic. However, recent reports of increasing minimal inhibitory concentrations of strains of S aureus to vancomycin have been associated with treatment failure. Vancomycin-resistant enterococci (VRE) also are being reported at an increasing rate, especially with hospital-acquired infections. Gram-positive bacilli, including Clostridium species, are very sensitive to vancomycin, but Gram-negative bacteria are resistant.
Vancomycin is not absorbed from the gastrointestinal tract. Maximum serum concentrations after a 10-mg/kg intravenous dose are approximately 25 mg/L, sixfold higher than the minimal inhibitory concentration of the usual bacteria being treated. It diffuses quite widely throughout the body and, during meningeal inflammation, attains concentrations in the cerebrospinal fluid approximately 10% to 20% of serum concentrations. However, penetration of vancomycin into epithelial lining fluid of the lung is quite variable, with concentrations averaging 25% of serum concentration. The t½ of vancomycin is approximately 4 to 6 hours in patients with normal renal function. The drug is excreted unmetabolized, almost exclusively in the urine. Doses should be reduced in patients with decreased renal function.
Vancomycin historically has had a reputation for toxicity. Many of its original adverse reactions, including ototoxicity and nephrotoxicity, were probably due to impurities in the formulation. Now that a more purified form is available, these adverse reactions are uncommon, although nephrotoxicity may occur with concomitant administration of an aminoglycoside. One of the more common side effects is the “red man” syndrome, which is characterized by fever, chills, erythema, and paresthesia. Although more likely to occur after a rapid infusion of vancomycin, “red man” syndrome also occurs after slow infusions and appears to be mediated by histamine.
Despite its introduction several decades ago, vancomycin recently has gained widespread use. The reasons for its revival relate to the emergence and increasing prevalence of several important pathogens. These include methicillin-resistant S aureus and coagulase-negative staphylococci, multiple-drug-resistant pneumococci, and enterotoxin-producing Clostridium difficile.
 AZTREONAM
AZTREONAM
Aztreonam is the first member of a unique class of antibiotics referred to as monobactams. Although monobactams are β-lactam antibiotics, their structure is so different that cross-immunogenicity does not appear to be a problem; they can be prescribed for patients with penicillin or cephalosporin allergies. Aztreonam is resistant to a broad range of β-lactamases produced by Gram-negative bacteria and therefore is active in vitro against most Gram-negative organisms. Activity against Gram-positive bacteria is limited. In comparison with the aminoglycosides, aztreonam appears to be less nephrotoxic and ototoxic. Clinical experience in children is limited.
 IMIPENEM AND MEROPENEM
IMIPENEM AND MEROPENEM
Imipenem and meropenem are members of the carbapenem β-lactam antibiotic family. Because imipenem is rapidly metabolized by renal brush-border enzymes, it is administered with cilastatin, a substance that inhibits imipenem metabolism by the kidney. Meropenem is administered alone, as it is more stable in vivo to inactivation by human renal dehydropeptidase. The carbapenems have activity against Gram-negative and Gram-positive aerobes and anaerobes. These antibiotics diffuse widely throughout the body and have excellent penetration into cerebrospinal fluid. They appear to have toxicity profiles similar to that of other β-lactam agents. Imipenem is epileptogenic in high doses, whereas meropenem appears to have less neurotoxicity. Experience with these antibiotics in children is limited, however carbapenems are useful in the treatment of extended-spectrum β-lactamase-producing Gram-negative bacteria that are resistant to all cephalosporins. In addition, they can be used as monotherapy for polymicrobial infections, which potentially include anaerobes such as intra-abdominal abscesses and necrotizing fasciitis.
 RIFAMPIN
RIFAMPIN
Rifampin is active against a wide range of Gram-positive and Gram-negative bacteria. It also is active against the majority of Mycobacterium tuberculosis strains, with minimal inhibitory concentrations less than 0.5 mg/L. Rifampin is given orally and is well absorbed from the gastrointestinal tract. Maximum serum concentrations of 8 mg/L are usually attained after a 600-mg dose. Rifampin penetrates well into most body tissues and fluids, including lungs, liver, pleural and ascitic fluid, bone, tears, saliva, and cerebrospinal fluid, even in the absence of inflammation. The t1/2 of rifampin ranges from 2 to 5 hours. It is metabolized in the liver and excreted principally in the bile and, to a lesser degree, in the urine.
Hypersensitivity reactions include dermatitis and a flulike syndrome, occasionally with thrombocytopenia, hemolytic anemia, and acute renal failure. Cholestatic hepatitis is another possible adverse reaction. All patients receiving this antibiotic should be advised that their bodily secretions, including urine, saliva, sweat, and tears, will develop a reddish-orange discoloration. This is especially important for patients who wear soft contact lenses, which may be permanently discolored.
Important drug interactions with rifampin have been recognized. For example, it enhances the metabolism of fluconazole, oral contraceptives, warfarin, propranolol, and anticonvulsants, all of which are metabolized in the liver. Doses of these concurrently administered agents may need to be increased to maintain therapeutic concentrations.
The use of rifampin as a single agent is limited by the fact that bacteria can rapidly develop resistance. It is, however, one of the first-line agents to be used in combination in the treatment of patients with most forms of tuberculosis. It also is the antibiotic of choice for the prophylaxis of contacts of patients with serious infections caused by H influenzae type b and N meningitidis. Rifampin also has been used to eradicate upper respiratory carriage of S aureus and group A streptococcus.
 QUINOLONES
QUINOLONES
The prototype of the quinolone antibiotics is nalidixic acid. This naphthyridine derivative has been used almost exclusively as a urinary antiseptic. It is as active as ampicillin against Gram-negative enteric isolates, but has no useful activity against Gram-positive bacteria or Pseudomonas species. Because nalidixic acid is only partially absorbed from the gastrointestinal tract, large doses are necessary to attain therapeutic urinary concentrations. These high doses have caused side effects, including visual disturbances. An additional problem has been the rapid development of bacterial resistance during therapy. These factors have limited the use of this antibiotic.
Research directed at modifying the chemical structure of nalidixic acid resulted in the development of the ever-growing family of fluorinated quinolone derivatives, including ciprofloxacin, ofloxacin, levofloxacin, gatifloxacin, and moxifloxacin. The spectrum of activity of these derivatives is continually increasing and now includes most Gram-positive bacteria, including some strains of methicillin-resistant S aureus and many Pseudomonas organisms. In addition, when compared with nalidixic acid, most Gram-negative enterics have greatly reduced minimal inhibitory concentrations to the new derivatives. Most quinolones are absorbed well after oral administration, and thus represent the first agents available for the oral treatment of systemic infections caused by resistant Gram-negative enteric isolates and Pseudomonas species. These agents are also of great value because their activity is unrelated to that of other antibiotics and resistance is not plasmid-borne. In adults, the quinolones may be preferred over alternate agents for treatment of complicated urinary tract infections, suspected bacterial gastroenteritis, osteomyelitis caused by Gram-negative bacilli, and invasive external otitis.
In animal studies, the quinolones caused cartilaginous damage in young animals. This limited their use in children to recalcitrant infections for which alternatives were lacking. However, recent data suggest that quinolones may be safe for administration to children, with the frequency of cartilage or joint toxicity being similar to that in adults. However, the American Academy of Pediatrics Committee on Infectious Diseases recommends that the use of quinolones should be limited in children because of high rates of resistance seen with overuse and the theoretical risk of joint injury.8 There are specific circumstances in which a quinolone may be the most appropriate antibiotic such as in urinary tract infections due to Pseudomonas species or other multidrug-resistant, Gram-negative bacteria or if other antibiotics are contraindicated because of allergy or resistance patterns.
 METRONIDAZOLE
METRONIDAZOLE
The antibacterial activity of metronidazole is limited to anaerobes, with greatest activity against Gram-negative anaerobic bacilli such as Bacteroides and Fusobacterium, most of which have minimal inhibitory concentrations under 3.12 mg/L. Activity against Gram-positive anaerobic cocci is less consistent, with about 75% of such strains being inhibited by 12.5 mg/L.
Metronidazole can be administered intravenously, orally, or rectally. Maximum serum concentrations after a 7.5-mg/kg dose administered intravenously are 20 to 25 mg/L. Concentrations after an equivalent oral dose are similar, and those after an equivalent rectal dose are about half. The drug diffuses well into all tissues; therapeutic concentrations can be attained in cerebrospinal fluid, bile, bone, and abscesses. The t1/2 of metronidazole is approximately 8 hours. It is metabolized to acid and hydroxy metabolites. Between 60% and 80% of the drug is eliminated by the kidneys, and 6% to 15% is eliminated in the feces. Hepatic insufficiency prolongs the t1/2of unchanged metronidazole, and doses usually have to be adjusted. Renal insufficiency usually does not necessitate dose adjustment.
Metronidazole therapy often is associated with a metallic taste and nausea. More serious but less frequent adverse reactions include a reversible peripheral neuropathy, seizures, encephalopathy, and neutropenia. A disulfiramlike reaction can occur when metronidazole is taken with alcohol. Several studies conducted in laboratory animals have indicated that prolonged use of high-dose metronidazole can be carcinogenic. However, there is no evidence that it is carcinogenic in humans.
Metronidazole has been shown to be effective in a wide variety of infections caused by anaerobes. The most common indications for this antibiotic are the treatment of pelvic and intra-abdominal sepsis and brain abscesses. It also is an effective and less expensive alternative to vancomycin in the treatment of pseudomembranous colitis caused by C difficile. Also, despite inconsistent in vitro activity of metronidazole against the principal etiologic agent of “nonspecific vaginitis,” Gardnerella vaginalis, it is the antibiotic of choice for treatment of this infection.
 AMINOGLYCOSIDES
AMINOGLYCOSIDES
The aminoglycoside group of antibiotics contains a large number of structurally related compounds. Streptomycin was the first of these agents to be discovered. Subsequently developed agents include neomycin, kanamycin, gentamicin, tobramycin, amikacin, and netilmicin. Streptomycin is primarily used to treat tuberculosis. Gentamicin, tobramycin, and amikacin are the most common aminoglycosides; they are discussed as a group, with only their clinically important differences emphasized.
These antibiotics are active primarily against Gram-negative and limited numbers of Gram-positive aerobes. They are inactive against the vast majority of anaerobes. All three of the aminoglycosides are active against most strains of P aeruginosa, with tobramycin consistently demonstrating the greatest activity. Gentamicin is consistently the most active of these agents against strains of Serratia marcescens. Otherwise, their relative antibacterial activities are similar, with most sensitive strains being inhibited by less than 3 to 4 mg/L.
An important aspect of aminoglycoside activity against Gram-negative aerobes is the increasing resistance developed since their introduction. Resistance is most often due to antibiotic inactivation by enzymes produced by the bacteria. There are at least 12 such inactivating enzymes. Gentamicin is susceptible to the largest number of these enzymes (9 of 12), and amikacin is susceptible to the smallest number (1 of 12). When widespread resistance develops to one of the aminoglycosides being used in a particular hospital, changing to an alternate agent usually results in a return to increased sensitivity.
The pharmacokinetics of all the aminoglyco-sides are similar. They are poorly absorbed from the gastrointestinal tract, but well absorbed after intramuscular or intravenous administration. Maximum serum concentrations of gentamicin and tobramycin are 5 to 8 mg/L after unit doses of 1 to 2.5 mg/kg. Maximum serum concentrations of amikacin range from 15 to 30 mg/L after a unit dose of 7.5 mg/kg. The aminoglycosides are distributed in most extracellular fluids, but do not attain therapeutic concentrations in cerebrospinal fluid. The main site of deposition of these drugs is the kidney, which accounts for approximately 40% of the total antibiotic in the body. The cortex accumulates approximately 85% of the load, and the resulting concentrations are more than 100-fold greater than serum concentrations. Their half-lives range from 1.5 to 2.5 hours, and they are eliminated, primarily unchanged, by glomerular filtration. The doses of the aminoglycosides must be carefully monitored and adjusted in the presence of renal insufficiency. The total daily dose is adjusted by either prolonging the dosing interval or reducing the unit dose. Nomograms, based on the measured or approximated glomerular filtration rate, are available to guide these adjustments.
Aminoglycosides demonstrate concentration-dependent killing, that is, the bactericidal activity increases with increasing concentration of drug. In addition, aminoglycosides exhibit a substantial postantibiotic effect. Aminoglycosides will inhibit growth of bacteria even after the serum level has fallen below the minimal inhibitory concentration for that antibiotic. Because of these pharmacodynamic characteristics, aminoglycosides may be effective when administered as a single daily dose. Toxicity is not increased using this dosing strategy.9
Toxicity
The most important toxicities of the aminoglycosides are ototoxicity and nephrotoxicity. These toxic effects are more common in adults than in children, who generally tolerate this class of drugs well. Ototoxicity may be primarily vestibular or cochlear. The agent most commonly associated with vestibular toxicity is gentamicin, with an estimated incidence in adult populations of 2%. This ranges from mild vertigo to severe Ménière syndrome. Damage is usually permanent, but symptoms may eventually be reduced by adaptation. The agents most likely to cause cochlear toxicity are amikacin and tobramycin. Although the frequency of hearing loss following treatment with these drugs is low, it may occur without any warning and may be irreversible. Risk factors that seem to predispose to ototoxicity include cumulative dosage, advanced age, and maternal history of preexisting renal compromise or hearing loss. Controlled trials in adult patients have found little difference in the incidence of ototoxicity following treatment with gentamicin, tobramycin, or amikacin.
Early manifestations of nephrotoxicity may include hypokalemia, glycosuria, alkalosis, hypomagnesemia, hypocalcemia, and enzymuria. The enzyme excreted as an early manifestation of aminoglycoside nephrotoxicity is the lysosomal enzyme N-acetyl-β-D-glucosaminidase (NAG). Renal damage is dose related and generally reversible.
Another less common but important side effect of the aminoglycosides is a competitive type of neuromuscular blockade, seen most often after intraperitoneal administration or after intravenous push. Hypersensitivity reactions to systemically administered aminoglycosides are uncommon.
Because of their relatively narrow toxic-to-therapeutic ratio, serum concentrations of the aminoglycosides should be monitored. When using multiple daily dosing, peak concentrations of gentamicin and tobramycin should not exceed 10 mg/L, and trough concentrations should be below 2 mg/L. Amikacin peak and trough concentrations should not exceed 30 mg/L and 10 mg/L, respectively. When using single daily dosing, levels approximately 8 hours after the start of dosing should be in the range of 2 to 5 mg/L for gentamicin and tobramycin and 10 to 15 mg/L for amikacin.
Indications
The most important indications for using one of the aminoglycosides are for treatment of proven or suspected Gram-negative infections of the blood, bones, joints, respiratory tract, urinary tract, or soft tissues. The aminoglycosides also are valuable in the empiric therapy of febrile, neutropenic episodes in immunocompromised patients.
 TETRACYCLINES
TETRACYCLINES
The tetracyclines are not frequently prescribed for children because of their age-related toxicities. The tetracyclines are active against a wide range of Gram-positive and Gram-negative bacteria, Mycoplasma, Rickettsia, and Chlamydia. They also are active against Treponema pallidum and moderately active against a wide range of anaerobes. All tetracyclines are absorbed adequately, but incompletely, from the gastrointestinal tract. They are chelated by various cations and are absorbed more completely during fasting. These antibiotics distribute widely and attain concentrations in the cerebrospinal fluid of 10% to 50% of simultaneous serum concentrations. Most of these agents are excreted primarily by renal glomerular filtration, with lesser amounts being eliminated in the bile. Doxycycline is an exception, with 90% appearing in the feces. The half-lives of the tetracyclines range from 6 hours for tetracycline to approximately 20 hours for doxycycline.
Toxicity
The adverse effects of tetracyclines relate to tooth and bone deposition. Permanent binding to dental calcium can produce a dose-related, brownish, fluorescent discoloration of the teeth when the drugs are administered during the period of dental calcification (from the fifth month of gestation to approximately age 8). Bone deposition may result in temporary cessation of bone growth. This effect is reversible when the drug is discontinued.
Other adverse effects of tetracyclines that are not age related include gastrointestinal disturbances, photosensitivity, hepatotoxicity, and neurotoxicity. Hypersensitivity reactions to the tetracyclines are rare.
Photosensitivity reactions may be caused by any of the tetracyclines but are most frequent with doxycycline. Unfortunately, doxycycline is frequently prescribed as a prophylactic agent against diarrhea in individuals traveling to tropical, sunny climates. Hepatotoxic reactions are uncommon, but fatal liver necrosis has been described after large intravenous doses in pregnant women. The pathogenesis of this reaction is unknown.
Manifestations of neurotoxicity are observed frequently and almost exclusively with minocycline. Dizziness, weakness, vertigo, and ataxia appear within the first few days of therapy. Another neurologic side effect of these agents is benign intracranial hypertension that is self-limited and resolves when the therapy is discontinued.
Indications
Indications for tetracycline therapy in adults and children over age 8 include infections caused by Mycoplasma pneumoniae, Q fever, psittacosis, brucellosis, rickettsial species, ehrlichiosis, and lymphogranuloma venereum. Tetracycline is also used to treat gonorrhea and syphilis in the penicillin-allergic nonpregnant patient and is frequently prescribed to patients with acne vulgaris. Doxycycline is an effective chemoprophylactic agent against E coli–induced diarrhea and against meningitis caused by N meningitidis or anthrax.
 LINEZOLID
LINEZOLID
Linezolid is the first member of the new oxazolidinone class of antibiotics. Linezolid binds to the 50S ribosomal subunit and prevents binding of the 30S subunit, mRNA, and initiation factors. It has 100% bioavailability after oral administration and has wide distribution throughout the body, including lung extracellular lining fluid and cerebrospinal fluid. Maximum serum concentrations of 11 to 16.7 μg/mL are achieved within 1 to 2 hours of an oral dose. Linezolid is cleared primarily by nonrenal mechanisms, with only 30% of the drug eliminated by the kidneys. The t1/2 is 1.5 to 5 hours; infants and young children clear the medication more rapidly.10 Linezolid has been approved for the treatment of complicated skin and skin-structure infections and nosocomial pneumonia. It demonstrates activity against Gram-positive bacteria such as staphylococcus, streptococcus, and enterococcus, including methicillin-resistant S aureus and vancomycin-resistant enterococcus (VRE).11 It also has been used as second-line therapy for both tuberculous and nontuberculous mycobacterium species.
Toxicity
Linezolid generally is well-tolerated in children. Most adverse effects occur only with prolonged (> 3 weeks) therapy. These include reversible hepatoxicity and bone marrow suppression. Linezolid is also a reversible monoamine oxidase inhibitor, so concurrent treatment with selective-serotonin reuptake inhibitors is contraindicated as this combination has been associated with serotonin-syndrome.
 CHLORAMPHENICOL
CHLORAMPHENICOL
Chloramphenicol is active against aerobic bacteria except P aeruginosa, most anaerobes, and the majority of Mycoplasma, Chlamydia, and Rickettsia organisms. Most susceptible bacteria have minimal inhibitory concentrations less than 5 mg/L.
Chloramphenicol is rapidly and completely absorbed from the gastrointestinal tract. The intravenous formulation of chloramphenicol is a succinate that must be hydrolyzed in vivo to biologically active free drug. Maximum serum concentrations attained after an oral or intravenous dose of 25 mg/kg range from 15 to 25 mg/L. There is, however, considerable inter-patient variability. Chloramphenicol diffuses well into most body fluids and tissues. Even in the absence of meningitis, concentrations in the cerebrospinal fluid often reach 70% to 80% of serum concentrations.
Chloramphenicol is metabolized in the liver. It is converted to a biologically inactive, water-soluble monoglucuronide. Impaired liver function can result in high serum concentrations. About 90% of chloramphenicol is excreted in the urine, but only 5% to 10% of this is in the unchanged biologically active form; dosage does not need to be adjusted in the presence of renal failure. The serum t1/2 is approximately 3 hours.
Toxicity
The most feared adverse effect of chloramphenicol therapy is aplastic anemia. This is not a dose-related phenomenon, and the mechanism is unclear. The precise frequency of this complication is not known but is estimated to be 1 in 40,000 treatment courses. A second type of hematopoietic depression is dose related. Serum concentrations in excess of 20 to 25 mg/L invariably result in reduced iron utilization by the bone marrow. This eventually leads to anemia and, less commonly, to thrombocytopenia and leukopenia. This type of marrow toxicity is reversible when the antibiotic is discontinued.
A toxic reaction to chloramphenicol in neonates is the “gray baby syndrome.” This is a form of circulatory collapse associated with excessive and sustained serum concentrations of unconjugated drug. Neonates are susceptible because of their immature hepatic drug-metabolizing enzymes.
Chloramphenicol serum concentrations should be monitored during therapy, and dosing should be adjusted if peak concentrations exceed 25 to 30 mg/L.
Indications
Chloramphenicol is effective in treating typhoid fever, rickettsial diseases, brain abscesses, and a variety of other infections in which anaerobes are usually pathogenic. In the penicillin-allergic patient, chloramphenicol is effective for infections caused by H influenzae, S pneumoniae, and N meningitidis. Because of the availability of equally effective, less-toxic agents, oral formulations of chloramphenicol are no longer made or available in the United States; an intravenous preparation is available.
 CLINDAMYCIN
CLINDAMYCIN
Clindamycin is active against most Gram-positive bacteria, both aerobic and anaerobic. It also is active against most Gram-negative anaerobic rods, but it is inactive against most Gram-negative aerobes. Sensitive organisms usually have minimal inhibitory concentrations less than 0.5 mg/L.
Clindamycin is well absorbed from the gastrointestinal tract. An oral dose of 300 mg results in maximum serum concentrations of 4 to 5 mg/L. It may be prescribed as a capsule or as a suspension. Maximum serum concentrations after an intravenous dose are two- to threefold higher than after an oral dose. Clindamycin distributes widely, but penetrates into cerebrospinal fluid poorly. The drug is metabolized primarily in the liver, with less than 25% of a dose ultimately excreted in the urine. Thus, hepatic insufficiency has a more profound effect on the disposition of this drug than does renal insufficiency. The t1/2 of clindamycin is 2 to 4 hours.
Toxicity
The most important group of adverse reactions to clindamycin are gastrointestinal disturbances. Approximately 30% of patients treated with this drug develop diarrhea. This diarrhea is usually self-limited and subsides when therapy is discontinued. It may be associated with nausea, vomiting, and abdominal cramps. A more severe gastrointestinal side effect is pseudomembranous colitis, which was first described in association with this antibiotic. It is caused by gastrointestinal overgrowth of toxin-producing C difficile. Almost every antibiotic has now been implicated in the pathogenesis of pseudomembranous colitis, and clindamycin is not the most frequent culprit. Furthermore, pseudomembranous colitis is much less common in children than in adults.
Minor abnormalities of liver function tests are quite common during clindamycin therapy, and cardiovascular collapse has been observed after rapid intravenous administration.
Indications
The most important uses of clindamycin are in treating a variety of anaerobic infections, including those caused by Bacteroides fragilis. Some infections treated successfully with clindamycin, usually combined with an aminoglycoside, include intra-abdominal and pelvic infections, aspiration pneumonia, infected decubitus ulcers, and periodontal disease. Recently, clindamycin has proved valuable in treating community-associated methicillin-resistant S aureus infections, as many of these isolates retain clindamycin susceptibility. Microbiology labs must ensure that inducible clindamycin resistance is not present by performing a D-test. Strains of methicillin-resistant S aureus that are resistant to erythromycin are more likely to demonstrate inducible resistance to clindamycin than strains that are sensitive to erythromycin. The presence of inducible clindamycin resistance has been associated with treatment failure.12 Clindamycin combined with penicillin is often recommended for treatment of necrotizing fasciitis due to group A streptococci.
 ERYTHROMYCIN
ERYTHROMYCIN
The antibacterial activity of erythromycin is similar to that of clindamycin. It is generally active against Gram-positive aerobes and anaerobes. It is inactive against most Gram-negative enterics but is active against certain nonenteric Gram-negative species, including Neisseria, Haemophilus, Bordetella, Campylobacter, and Legionella. The Gram-negative anaerobes are not reliably sensitive. Rickettsia, Mycoplasma pneumoniae, Ureaplasma, and Chlamydia are usually inhibited by attainable concentrations of erythromycin. Most sensitive bacteria are inhibited by less than 1.0 mg/L of this antibiotic.
Erythromycin base is adequately absorbed from the gastrointestinal tract. The base is inactivated by gastric acidity, and therefore absorption can be enhanced by enclosing the antibiotic in a capsule or by administering it as a stearate or estolate derivative. Maximum serum concentrations after a 500-mg dose of base or stearate are approximately 1 mg/L. Concentrations are twoto fourfold higher after an equivalent dose of the estolate formulation. A 500-mg dose of intravenous erythromycin results in maximum serum concentrations of about 5 mg/L.
Erythromycin is distributed throughout body water. It attains only low concentrations in the cerebrospinal fluid, even with inflamed meninges. Only a small amount of erythromycin is excreted in its original form; the remainder is metabolized. The t1/2 is approximately 2 hours.
Toxicity
Oral erythromycin formulations often result in gastrointestinal disturbances, including nausea, vomiting, diarrhea, and abdominal cramps. These adverse effects are likely to occur at high doses. A much more serious adverse reaction, fortunately rare among children, is cholestatic hepatitis. It occurs most commonly with the estolate preparation and is probably due to the propionyl ester linkage. Manifestations can include jaundice, fever, pruritus, rash, increased liver size, and eosinophilia. Resolution usually occurs when the antibiotic is discontinued. Erythromycin use in neonates has been associated with the development of pyloric stenosis.
Intravenous erythromycin is frequently associated with thrombophlebitis. Ototoxicity, manifested as tinnitus and transient deafness, is a rare adverse reaction.
Indications
Erythromycin is an effective alternative to penicillin for treating streptococcal and pneumococcal infections, although many S pneumoniae are becoming resistant. Erythromycin also is indicated for treating respiratory Mycoplasma infections; for eradicating Bordetella pertussis and Corynebacteria diphtheria from the nasopharynx, Chlamydia infections, Legionnaire’s disease, and gonorrhea or syphilis during pregnancy; and for eradicating Campylobacter from the stools of patients with Campylobacter gastroenteritis. Erythromycin should not be used alone in treating otitis media. Although it is active in vitro against the majority of bacteria responsible for this infection, middle ear concentrations are not consistently above the minimal inhibitory concentration for strains of H influenzae. If used for this indication, it should be given with a sulfonamide. An erythromycin-sulfonamide fixed combination is marketed for this indication.
 AZITHROMYCIN
AZITHROMYCIN
Azithromycin is an azalide antibiotic that is structurally related to erythromycin. Its biochemical modifications result in superior oral bioavailability, a greatly extended serum and tissue half-life (both exceeding 48 hours), and excellent in vivo activity against most of the organisms susceptible to erythromycin. In addition, it has excellent activity against Chlamydia trachomatis, with minimal inhibitory concentrations between 0.03 and 0.5 mg/L. It is particularly well suited for treating genital infections caused by Chlamydia. A single oral dose is as effective as a 7-day course of erythromycin or doxycycline.
 CLARITHROMYCIN
CLARITHROMYCIN
Clarithromycin is another macrolide antibiotic that is similar to azithromycin. A special feature of clarithromycin is its activity against selected mycobacteria. It is particularly useful in the treatment of atypical mycobacteria infections, especially those caused by Mycobacterium avium-intracellulare.
 SULFONAMIDES
SULFONAMIDES
Sulfonamides were the first group of synthetic antibacterial compounds. These antibiotics originally had a wide range of activity, but this range is considerably compromised by acquired bacterial resistance. Gram-positive bacteria that are usually sensitive to sulfonamides include group A streptococci, Streptococcus viridans, some S pneumoniae, and Nocardia species. Staphylococci are variably sensitive, and Streptococcus faecalis is resistant. The most sensitive Gram-negative bacteria are Neisseria species, many enterobacteria, H influenzae, and B pertussis. Chlamydia and nonbacterial pathogens such as Toxoplasma and Plasmodium falciparum are also sensitive to the sulfonamides.
The sulfonamides are often classified on the basis of their half-lives, which range from 2 to 6 hours with the short-acting sulfonamides, such as sulfanilamide, sulfadiazine, and sulfisoxazole, to 150 to 200 hours with the ultralong-acting sulfonamide sulfadoxine. Most of the sulfonamides are well absorbed from the gastrointestinal tract. Serum concentrations vary somewhat among the different agents, but after the usual recommended, orally administered doses, maximum concentrations are typically in the range of 50 to 100 mg/L. Concentrations are higher after intravenous administration. These antibiotics are distributed widely and attain therapeutic concentrations in cerebrospinal fluid. The sulfonamides are acetylated in the liver, and some also undergo glucuronidation. Free and conjugated sulfonamides are excreted by renal glomerular filtration and secretion. The longer-acting sulfonamides undergo more complete tubular resorption than do the shorter-acting agents. Minimal amounts of the sulfonamides are excreted in the bile.
Toxicity
Sulfonamides may cause a variety of hypersensitivity reactions, ranging from mild rashes to life-threatening Stevens-Johnson syndrome. The latter reaction is more common with the longer-acting sulfonamides. Hematologic toxicity also may occur with sulfonamide use. Reactions include agranulocytosis, which is usually reversible on discontinuation of the drug, and hemolytic anemia in patients with deficiency of G6PD. Renal damage was common with the older sulfonamides, which were poorly water soluble. Patients developed crystalluria, which led to urinary obstruction and hematuria. Renal damage may be a manifestation of a hypersensitivity reaction. Sulfonamides are contraindicated in the neonate and during the latter part of pregnancy, as they may displace bilirubin from protein-binding sites, possibly leading to jaundice and kernicterus. Neonates seem to be more susceptible to the potential renal toxicity of these agents.
Indications
Clinical uses of the sulfonamides include the treatment of acute, uncomplicated urinary tract infections and infections caused by Chlamydia, Nocardia, Toxoplasma, and chloroquine-resistant P falciparum. For the latter two pathogens the sulfonamide is administered combined with pyrimethamine. The sulfonamides are also used as prophylactic agents; for example, in children with rheumatic fever who are allergic to penicillin and in children with frequently recurring urinary tract infections. When used to reduce the incidence of recurrent urinary tract infections, the sulfonamide is usually administered in combination with trimethoprim.
 TRIMETHOPRIM
TRIMETHOPRIM
Trimethoprim has a bacterial spectrum similar to that of the sulfonamides, although it generally has lower minimal inhibitory concentrations against most isolates. Trimethoprim is active against enterococci, whereas the sulfonamides are not.
Trimethoprim is well absorbed from the gastrointestinal tract. Maximum serum concentrations of 2 mg/L are attained after a 160-mg dose. Tissue concentrations of this antibiotic often exceed serum concentrations except in the brain, skin, and fat. Trimethoprim is metabolized primarily in the liver. Approximately 50% of an administered dose is excreted unchanged in the urine, and the remainder is excreted as metabolites. The t1/2 is about 11 hours.
At high dosages, trimethoprim may cause nausea and vomiting. Blood dyscrasias have occurred rarely. Because trimethoprim is an antifolate, anemia secondary to folate deficiency may occur, especially among patients with a preexisting folate deficiency.
Trimethoprim is commonly used with another antibiotic, usually a sulfonamide. Infections treated with this combination include urinary tract infections, sinusitis, otitis media, shigellosis, nocardiasis, and Pneumocystis carinii pneumonitis. Systemic infections caused by Gram-negative aerobes resistant to multiple antibiotics have also been treated with this antibiotic combination. In addition, the combination is effective prophylactically in patients with recurrent urinary tract infections and in immunocompromised patients at risk for pneumonia caused by P carinii.
 NITROFURANTOIN
NITROFURANTOIN
Although nitrofurantoin was approved by the Food and Drug Administration in 1953, its exact mechanism of action is still not known. It has a hydantoin ring with a nitro-substituted furanyl side chain that is metabolized by bacteria to activate its bactericidal activity.4 Nitrofurantoin has broad activity against Gram-positive and Gram-negative enteric bacteria.
Nitrofurantoin is well absorbed after oral administration and rapidly cleared by the kidneys. Therefore, serum levels are not maintained, but the antibiotic concentrates in the urine, making it a useful antibiotic for urinary tract infections. Nitrofurantoin has an extremely short half-life (∼30 min) and a high volume of distribution, which may be due to both rapid distribution into tissue compartments and enzymatic degradation at those sites.
Toxicity
The primary side effect of nitrofurantoin therapy is nausea and vomiting. These gastrointestinal adverse effects are the most common reason for discontinuing therapy. Although there have been reports of acute lung injury and pulmonary fibrosis in adults on long-term nitrofurantoin therapy, similar toxicities have not been seen in children. Hemolysis can occur in patients with glucose-6-phosphate dehydrogenase deficiency.
Indications
Because of the high concentrations of nitrofurantoin in urine and its broad-spectrum of activity, it has primarily been used as both therapy and prophylaxis in urinary tract infections (UTIs). Some data suggest that nitrofurantoin may be more effective than other antibiotics in preventing recurrent UTIs in children. However, its gastrointestinal side effects may limit its utility.13
SPECIAL ISSUES IN PEDIATRIC ANTIBIOTIC THERAPY
 INTRAVENOUS INFUSIONS
INTRAVENOUS INFUSIONS
Antibiotics frequently are given intravenously to hospitalized children with serious infections. Intravenous administration does not ensure drug delivery. Antibiotic stability in the delivery solution and potential drug incompatibilities must be considered. As a general rule, intravenous drugs should always be given separately. If this is not possible, the compatibility of the mixed agents must be verified.
When drugs are given intravenously, the delivery system itself must also be considered. Pediatric unit doses may be so small relative to the volume in the intravenous infusion system that it becomes impossible to determine whether the dose has been completely delivered. The use of dilute solutions of the antibiotics should circumvent this problem. The use of in-line filters with infusion systems may remove certain antibiotics. In addition, certain drugs may adhere to the plastics of the infusion sets, reducing the dose delivered to the patient.
 INFLUENCE OF FOOD AND BEVERAGES ON ORAL ANTIBIOTICS
INFLUENCE OF FOOD AND BEVERAGES ON ORAL ANTIBIOTICS
Major factors in achieving compliance in young children are smell and palatability. Table 246-7 indicates which antibiotics should be taken on an empty stomach and which should be taken with food.
Table 246–7. Antibiotic Administration and Food Consumption



