Anemia in the Neonatal Period
INTRODUCTION
The newborn period marks a time when red blood cell (RBC) indices change significantly. Anemia can occur at various times in the neonatal period, from the perinatal and immediate postnatal period through the first months of life. Hematocrits 2 or more standard deviations below the normal range for gestation are seen frequently and should be evaluated. Conversely, true anemia, the inability to adequately deliver oxygen to tissues, is less common. Anemia can be classified into the following 3 major processes: hemolysis, hemorrhage, or hypoproliferative disease. Anemia can also result from overlapping processes. For example, sepsis can result in hemolysis, disseminated intravascular coagulation (DIC), and subsequent hemorrhage. This chapter reviews fetal and neonatal erythropoiesis, discusses the etiology and diagnosis of anemia in the neonatal period, and offers management options for anemic term and preterm infants.
Fetal Erythropoiesis
The growth factors stimulating fetal erythropoiesis during gestation are produced solely by the fetus. Erythropoietin, or Epo, is the primary growth factor responsible for erythropoiesis after birth and appears to be the principal growth factor for fetal erythropoiesis as well. Primary production of Epo occurs in the liver during fetal development. The kidney becomes the primary source of Epo following delivery; however, the etiology for the liver-to-kidney switch continues to be studied. Anephric fetuses have normal serum Epo concentrations and normal hematocrits, proving that renal Epo production is not required for erythropoiesis during fetal development. Methylation patterns in the promoter and enhancer regions of the Epo gene isolated from fetal liver and kidney suggest increased methylation in the kidney as a possible explanation for decreased Epo gene expression during fetal development.
During gestation, the site of red cell production transitions from yolk sac to liver to marrow. Primitive erythroblasts are produced in the fetal yolk sac during the first 3–4 weeks of gestation. By 6 to 8 weeks’ gestation, definitive erythropoiesis is established in the fetal liver. Although erythrocytes are noted in the fetal marrow as early as 8–9 weeks’ gestation, the liver remains the primary site of erythropoiesis until well into the second trimester. By the third trimester, erythropoiesis occurs primarily in the fetal marrow, although production can continue in extramedullary sites in the face of increased need, during times of hemolysis, or in fetal infection caused by a variety of bacteria or viruses.
Variations in hematologic indices for preterm and term infants reflect the dynamic nature of the erythron in late fetal development. The production of hemoglobin transitions from embryonic (Gower 1 [ζ2ε2], Gower 2 [α2ε2], and Portland [ζ2γ2]) to fetal hemoglobin (α2γ2) and finally to adult hemoglobin (α2β2), as shown in Figure 31-1.
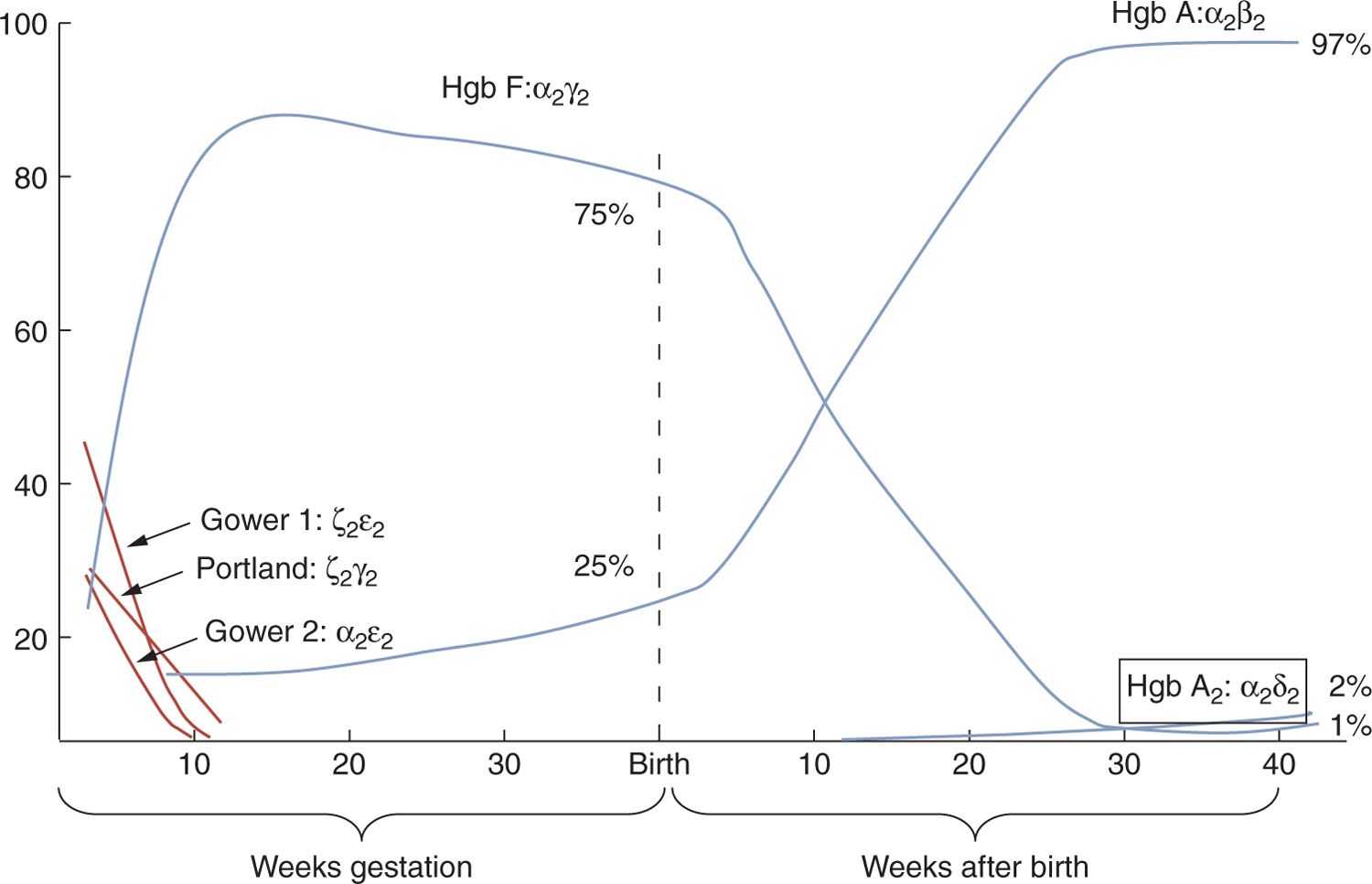
FIGURE 31-1 Changes in hemoglobin production from conception through 40 weeks postnatal age.
In conjunction with changes in globin gene expression and the transition from embryonic to fetal hemoglobin production, hemoglobin concentrations gradually increase, from approximately 14.5 g/dL at 28 weeks’ gestation to 15 g/dL at 34 weeks to 16.8 g/dL at 40 weeks.1 The relationship between cord hemoglobin concentration and duration of pregnancy is linear for infants who are appropriate for gestational age (AGA). The mean corpuscular volume (MCV) is higher in preterm infants than in term infants and is inversely proportional to gestation, averaging 5–20 fL higher than the mean MCV of 108 fL in term infants.
Reticulocyte counts are elevated at birth, averaging 6% to 10%. Nucleated red blood cells (NRBCs) are not consistently elevated, but there does appear to be an inverse relationship between number of NRBCs and gestational age. Normoblasts are cleared rapidly from the circulation during the first postnatal days, although a few may still be observed in small preterm infants into the second week of life. Variations in red cell size and shape are somewhat greater than those observed in term infants.
Neonatal Erythropoiesis
The abrupt transition from the relatively hypoxic in utero environment to the oxygen-rich postnatal environment triggers erythropoietic responses that have a profound effect on the red cell mass. Over the first 2 to 6 months of life, the infant experiences both the highest and the lowest hemoglobin concentrations occurring at any time in development. Although variable, Epo levels at birth are above the normal adult range but decrease abruptly in the immediate postnatal period, with a half-life of no more than 4 hours (2.6 ± 0.5 hours in infants with polycythemia and 3.7 ± 0.9 hours in infants born to mothers with preeclampsia.)2 By 24 hours, the Epo value is nearly undetectable and remains so throughout the first month of life. The decrease in Epo is accompanied by a decrease in the number of erythroid progenitors and in the reticulocyte count.3
A combination of decreased red cell production, shortened cell survival, and growth-related expansion of the blood volume results in a progressive fall of the hemoglobin concentration to a mean of approximately 11 g/dL at 2–3 months of age.1 The lower range of normal for infants of this age is approximately 9 g/dL. This nadir is called physiologic anemia, in that it is not associated with lack of oxygen delivery to tissues and is not abrogated by nutritional supplements. Stabilization of the hemoglobin concentration is heralded by an increase in reticulocytes in the second month of life. Thereafter, the hemoglobin concentration rises to an average of 12.5 g/dL, where it remains throughout infancy and early childhood.
ETIOLOGY
Hemolysis
Hemolysis is the premature destruction of erythrocytes and is generally categorized as immune-mediated or non-immune-mediated hemolysis (Table 31-1). Immune-mediated hemolysis occurs when immunoglobulin (Ig) G antibodies created by the maternal immune system cross the placenta and cause destruction of antigen-containing fetal red cells (isoimmunization). Hemolysis in the fetus can be severe enough to lead to hydrops fetalis. Non-immune-mediated hemolysis occurs when intrinsic abnormalities in the fetal or neonatal red cell lead to premature destruction and a shortened red cell life span.
Table 31-1 Differential Diagnosis of Hemolysis in the Fetal and Neonatal Period
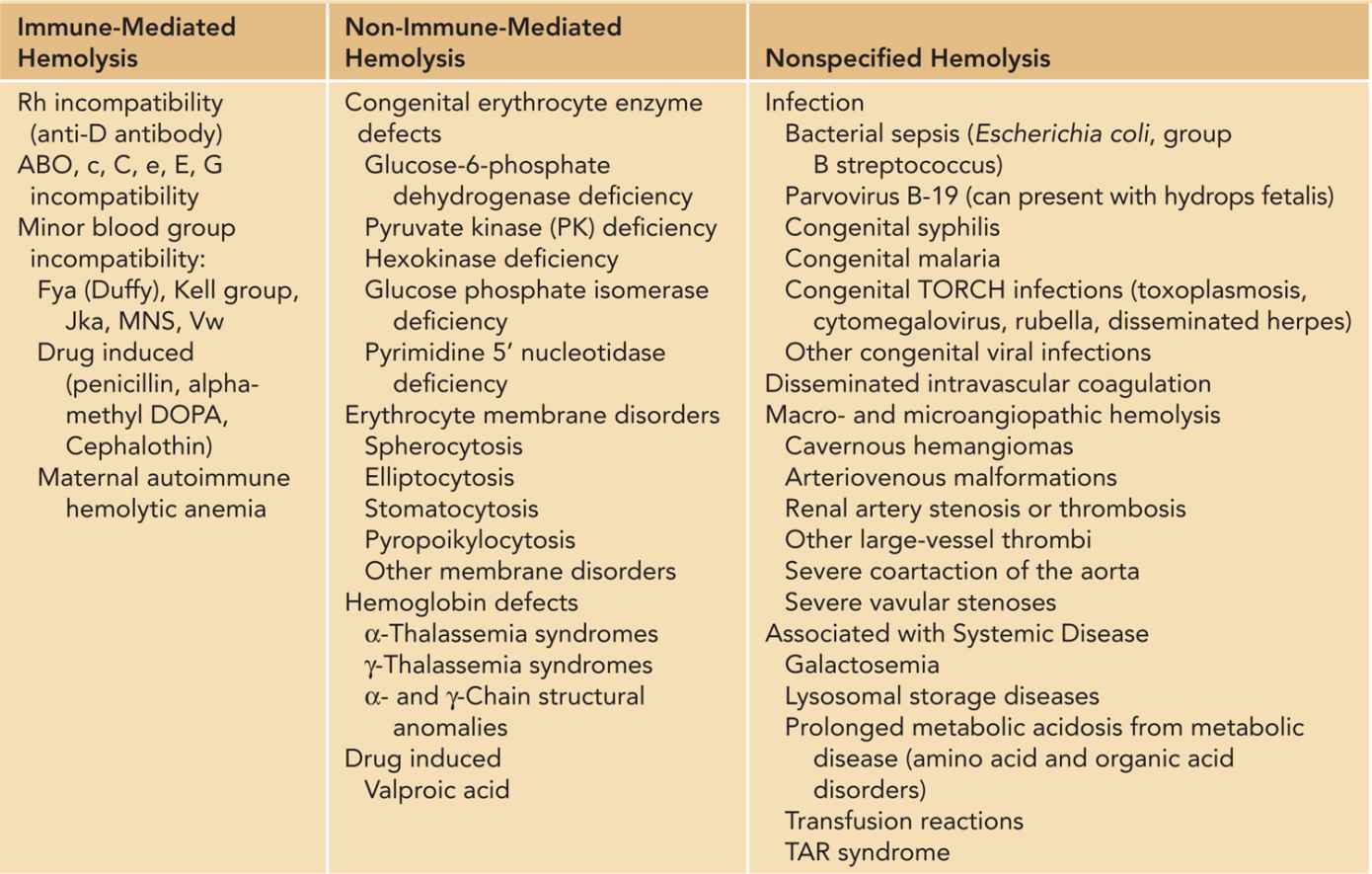
In older children and adults with congenital hemolytic anemia, there is usually clear evidence of hemolysis, as manifested by indirect hyperbilirubinemia, reticulocytosis, and low haptoglobin levels, all markers of increased red cell destruction. In neonates, however, the recognition of congenital hemolytic anemia utilizing these laboratory measurements is more challenging. Physiologic indirect hyperbilirubinemia is common in neonates because of the increased red cell mass at birth, decreased RBC survival, and reduced hepatic glucuronyl transferase activity. Also, the normal physiologic anemia of infancy can modulate reticulocytosis in the presence of mild hemolytic anemia. Finally, serum haptoglobin is not a reliable index of neonatal hemolysis because concentrations of this protein do not increase until after 6 months of age.
Neonates with congenital hemolytic anemia may have a normal-for-age hemoglobin or there may be mild-to-severe anemia. Indirect hyperbilirubinemia presenting earlier than expected and in excess of the usual physiologic levels is the most common and occasionally only sign of hemolytic anemia. Reticulocytosis and persistence of nucleated RBCs beyond the third day of life are other reasons to evaluate a neonate for hemolytic anemia. Acquired hemolytic anemia in neonates occurs with alloimmune hemolysis (chapter 34), hemolytic anemia secondary to maternal disease, and hemolysis secondary to infection or microangiopathic anemia.
Immune-Mediated Hemolysis
The most common cause for hemolysis in the neonatal period is immune-mediated hemolysis due to ABO incompatibility or Rh incompatibility. Following the advent of RhoGAM (RhD antibody), the incidence of hydrops fetalis due to RhD sensitization decreased significantly, from approximately 1 in 200 live births to now less than 1 in 1000 live births.4 Immune-mediated hemolysis due to anti-A or anti-B antibodies crossing the placenta is now the most common etiology of immune-mediated hemolysis.
RhD Isoimmunization: RhD incompatibility occurs when a woman with RhD-negative blood type is exposed to Rh-positive blood cells, leading to the development of RhD antibodies (anti-D antibodies). Fifteen percent of the population lacks the D antigen (is Rh negative). RhD sensitization occurs in approximately 1 per 1000 births to women who are Rh negative.
RhD incompatibility occurs through 2 primary mechanisms. The most common route of exposure occurs when an Rh-negative pregnant mother is exposed to Rh-positive fetal RBCs secondary to fetomaternal hemorrhage (FMH), spontaneous abortion, or normal delivery. RhD incompatibility can also occur if an Rh-negative female accidentally receives Rh-positive blood. Maternal anti-D IgG antibodies freely cross the placenta into the fetal circulation, where they form antigen-antibody complexes with RhD-positive fetal RBCs. The red cells are eventually lysed, leading to isoimmune hemolytic anemia.5 As a result, large amounts of bilirubin are produced from the breakdown of fetal hemoglobin and are transferred via the placenta to the mother, where they are conjugated and excreted.
Once the baby is born, low levels of glucuronyl transferase can lead to significant elevations of unconjugated hyperbilirubinemia. Erythroblastosis fetalis is a severe form of the disease characterized by severe hemolytic anemia and hydrops fetalis, involving placental and fetal edema, ascites, pericardial effusion, high-output cardiac failure, and extramedullary hematopoiesis. The risk and severity of sensitization increase with each subsequent pregnancy in which the fetus is Rh positive. The risk of sensitization depends on the volume of FMH, the extent of maternal immune response, and the concurrent presence of ABO incompatibility, which actually decreases the severity of hemolysis in Rh-positive fetuses.
Administration of RhoGAM decreases the production of maternal IgG antibodies by inhibiting immune memory. RhD immune globulin coats the surface of RhD-positive fetal RBCs. These antibody-antigen complexes inhibit the stimulation of maternal B cells to produce IgG antibodies. RhoGAM is administered once at 28–32 weeks’ gestation. It has a short half-life of 23–28 days and is thus administered again within 72 hours after delivery. Rh-negative (nonsensitized) women presenting to the emergency room with antepartum bleeding or potential FMH should receive 300 μg of Rh IgG.6 An additional 300 μg of Rh IgG should be administered for every 30 mL of fetal blood to which the Rh-negative mother is exposed. A lower 50-μg dose (MICRhoGAM) is available and recommended for Rh-negative women who have a termination of pregnancy in the first trimester when the volume of FMH is likely minimal.7
ABO Isoimmunization: Hemolysis of fetal red cells caused by ABO antibodies occurs almost exclusively in infants of blood group A or B who are born to group O mothers.8 Anti-A and anti-B antibodies formed in group O individuals are mainly IgG antibodies and thus can cross the placenta, whereas anti-A and anti-B antibodies found in the serum of group A or group B individuals tend to be IgM antibodies. ABO incompatibility tends to be relatively mild in nature, mainly because fetal RBCs do not express adult levels of A and B antigens, resulting in few reactive sites. This allows the antibody-coated cells to remain in the circulation for a longer period of time compared to RhD disease. In addition, severity of ABO incompatibility is not dependent on birth order.9
The presence of microspherocytes on peripheral blood smear is a characteristic of ABO isoimmunization, with little or no increase in nucleated RBCs. In RhD isoimmunization, there are fewer spherocytes and a large number of nucleated RBCs.10 ABO isoimmunization usually presents with hyperbilirubinemia and can be managed by phototherapy alone. Exchange transfusions are occasionally required. The direct antiglobulin test (DAT) is usually weakly positive but can be negative in some cases despite evidence of immune-mediated hemolysis.
Minor Blood Group Isoimmunization: Although RhD isoimmunization is the most common cause of severe hemolytic disease of the newborn, other antibodies belonging to Kell (K and k), Duffy (Fya), Lewis, Kidd (Jka and Jkb), and MNSs (M, N, S, and s) systems can cause severe hemolytic disease of the newborn.11 Kell antigen is a minor blood group antigen that often results in isoimmunization. In addition, antibodies directed against Kell antigens can bind to erythroid progenitors in the fetal marrow and result in hypoproliferative anemia.12 Minor Rhesus blood group antigens c and E can also cause severe hemolysis, requiring exchange transfusion.
Non-Immune-Mediated Hemolysis
Enzyme Disorders
Glucose is the major metabolic substrate for RBCs, and it is metabolized via the glycolytic pathway and hexose monophosphate (HMP) shunt. The glycolytic pathway generates adenosine triphosphate (ATP), NADH (reduced nicotinamide adenine dinucleotide), and 2,3-disphosphoglycerate (2,3-DPG), all important in maintaining RBC integrity. The HMP shunt pathway is critical for protecting RBCs against oxidative insult. Glucose-6-phosphate dehydrogenase (G-6-PD) is the initial step in the HMP pathway. Among hemolytic anemias due to hereditary enzyme deficiencies, G-6-PD deficiency is the most common. The remaining glycolytic pathway enzymopathies are relatively rare, with only a few thousand cases known and 90% due to pyruvate kinase deficiency.
Glucose-6-Phosphate Dehydrogenase Deficiency: Deficiency of G-6-PD is one of the most common genetic disorders in the world, affecting over 400 million people worldwide.13 It is a sex-linked disorder affecting males, but females who are homozygous for G-6-PD deficiency or have skewed lyonization of the X chromosome can also have significant enzyme deficiency. This enzymopathy is found most commonly in people from Africa, Asia, and the Mediterranean region. Three clinically important types of G-6-PD deficiency are classified by the World Health Organization (WHO). Class I G-6-PD deficiency is vanishingly rare, but it is the most severe form, characterized by lifelong mild-to-moderate chronic hemolytic anemia. Class II G-6-PD variants, commonly found in Mediterranean (G-6-PDMediterranean), Middle Eastern, and Asian people, have a marked enzyme deficiency that is associated with severe anemia after exposure to oxidant drugs and chemicals (eg, primaquine, ciprofloxacin, nitrofurans, sulfonamides, dapsone, naphthalene) or fava beans. Class III G-6-PD is the mildest deficiency, found in people of African (A-variant) and Asian background who experience moderate but self-limited hemolytic anemia with oxidant exposure. In patients with class II or III G-6-PD, there is no anemia or hemolysis in the steady state when there is no oxidant exposure.
G-6-PD deficiency in neonates is associated with high risk of severe jaundice and kernicterus.14,15 It is much more common in neonates with class II variants than in infants with class III G-6-PD deficiency. The possibility of G-6-PD deficiency should be considered in infants with hyperbilirubinemia beyond expected physiologic levels. The neonatal jaundice associated with G-6-PD deficiency has a late peak at day 5–6 of life; it may be especially difficult to identify in preterm infants due to their later peak bilirubin levels. Of particular importance, hyperbilirubinemia often is the only abnormality, and there may be no other signs of hemolysis. This observation has suggested that hyperbilirubinemia might not be related solely to increased bilirubin production from red cell destruction but rather may be due to a defect in liver clearance of bilirubin.16 Also, there are reports suggesting that marked hyperbilirubinemia in G-6-PD deficiency is associated with the simultaneous inheritance of the variant Gilbert polymorphism.17 In the United States, severe hyperbilirubinemia due to G-6-PD deficiency is responsible for over 30% of kernicterus cases.18 Neonates at risk for G-6-PD should have bilirubin levels measured prior to discharge. Close outpatient follow-up is particularly important given that many neonates are discharged early from the hospital. Neonatal discharge guidelines have been established by the American Academy of Pediatrics and the American College of Obstetricians and Gynecologists.19
Severe hyperbilirubinemia occurring in G-6-PD deficiency requires phototherapy and, occasionally, exchange transfusion. Packed RBC (PRBC) transfusion should be given for symptomatic anemia. Avoidance of oxidative medications and chemicals is important to prevent further hemolysis. Mothers who are breastfeeding should be counseled to avoid ingestion of fava beans and oxidant medications. Vitamin K injection is safe for neonates with class II and III variants and should be given to prevent hemorrhagic disease of the newborn.
Pyruvate Kinase Deficiency: Pyruvate kinase (PK) deficiency is the most common glycolytic pathway defect. Over 200 different mutations of the 2 different PK genes (PKM2 and PK-LR) have been reported.20 It is an autosomal disorder, with most patients compound heterozygous for 2 different mutations. The prevalence of PK deficiency among Caucasians in the United States has been estimated at 1 in 20,000. The disease is common among the Amish population in Pennsylvania and other inbred communities.21
Pyruvate kinase catalyzes the conversion of phosphoenolpyruvate to pyruvate, at the same time generating ATP, a critical source of energy for RBCs. Impaired ATP production occurs in PK-deficient cells, leading to loss of membrane plasticity and, ultimately, splenic destruction of red cells.
The clinical features of PK deficiency are highly variable, ranging from severe anemia and jaundice at birth to an incidental finding in adults. This is likely related to the many different known mutations and the often-compound heterozygosity for different mutations causing the deficiency.
Deficiency of PK should be part of the differential diagnosis of hydrops fetalis and neonatal jaundice. Jaundice can be pronounced, and kernicterus has been reported.22 In contrast to G-6-PD deficiency, in addition to hyperbilirubinemia, there are usually obvious features of hemolysis, including anemia and reticulocytosis. PK deficiency should be suspected in a neonate with evidence of hemolytic anemia after immune-mediated hemolysis, hereditary spherocytosis (HS), and G-6-PD deficiency are ruled out.
Hyperbilirubinemia should be managed with phototherapy and exchange transfusion as needed. Severe anemia may require red cell transfusions to maintain hemoglobin or as needed to maintain normal neonatal growth and development. Many children with severe PK deficiency are transfusion dependent until they undergo splenectomy after 5 years of age.
Other RBC Enzymopathies Associated With Hemolysis: Abnormalities have been reported in all glycolytic enzymes. These conditions are rare, accounting for less than 1% of hemolytic anemia due to enzymopathies.23 After PK deficiency, which accounts for 90% of glycolytic enzymopathies, glucose phosphate isomerase (GPI) deficiency is the next most common. All glycolytic enzymopathies are associated with chronic nonspherocytic hemolytic anemia. The degree of hemolytic anemia varies from mild to severe, and hydrops fetalis has been seen in those with the most severe hemolysis. Several glycolytic enzyme deficiencies are associated with myopathy or neurologic deficits. Most are inherited in an autosomal recessive (AR) pattern except phosphoglycerate kinase deficiency, which is X linked. This group of rare disorders should be considered when a neonate shows clear and persistent evidence of nonspherocytic hemolytic anemia and after other hemolytic causes such as immune-mediated hemolysis, membrane defects, and the more common enzymopathies discussed have been ruled out. Enzymopathies should also be considered in neonates with neurologic defects or myopathy in addition to hemolytic anemia. Specific assays of RBC enzyme activity are necessary for diagnosis of these rare conditions. In some cases, DNA-based molecular tests are available.
Membrane Disorders
Hemolytic anemias due to abnormalities of the erythrocyte membrane comprise an important group of inherited disorders. The most common disorder encountered is HS. Hemolysis due to hereditary elliptocytosis (HE), and the hereditary stomatocytosis (HSt) syndromes also occurs but is less common.
Hereditary Spherocytosis: Hereditary spherocytosis is the most common hereditary hemolytic anemia in people of northern European descent, occurring in up to 1 in 2500 individuals.24 In the United States, it is estimated that 1 in 5000 people have HS. Most cases of HS are inherited in an autosomal dominant (AD) pattern. However, up to 25% of cases do not display this inheritance; some are new mutations, while others represent AR inheritance.
Hereditary spherocytosis is associated with deficiencies of membrane cytoskeleton proteins (spectrin, ankyrin, or band 3) that lead to destabilization of the RBC lipid bilayer, resulting in progressive membrane surface loss and microspherocyte formation.25 Microspherocytes are vulnerable to entrapment by the spleen, which leads to further membrane loss and ultimately cell destruction by splenic macrophages. A characteristic feature of HS is the presence of spherocytes seen in the peripheral blood smear. However, even normal neonates can have a small population of spherocytes, and as noted previously, spherocytes also are found in neonates with hemolysis due to ABO incompatibility. Older children and adults with HS commonly have anemia, reticulocytosis, indirect hyperbilirubinemia, and splenomegaly.
In neonates, the most common presentation of HS is jaundice. Jaundice may present early, be more severe than usual, persist longer, and may require treatment with phototherapy. Infants with HS rarely require an exchange transfusion. Up to 50% of adults with HS have a history of jaundice during the first week of life.26 Those who coinherit HS and the gene for Gilbert syndrome may develop marked jaundice requiring phototherapy.
The diagnosis of HS can be made in a neonate with a positive family history of HS who has evidence of hemolytic anemia, the presence of an increased number of spherocytes on peripheral smear, elevation of mean corpuscular hemoglobin concentration (MCHC), and a negative DAT. In these cases, further testing is not necessary. However, if the family history is in doubt or if there is coexistence of ABO incompatibility, further testing of the neonate or repeat testing of the affected family member might be necessary to confirm the diagnosis. The incubated RBC osmotic test is used to detect spherocytosis. However, it is important to recognize that this test does not differentiate the various causes of spherocytosis. If there is an ABO incompatibility setup, it is important to obtain a DAT. Occasionally, the establishment of the diagnosis of HS needs to be delayed until confounding factors (maternal antibodies, fetal RBC changes) are resolved. Since management and counseling of neonates with hemolytic anemia is not dependent on the exact cause, the complete laboratory workup can usually be deferred for 4 to 6 months.
Occasionally, severe symptomatic anemia can develop soon after birth and require a PRBC transfusion. More often, anemia develops after discharge from the nursery. It therefore is important to have close follow-up of these infants. While some neonates do not develop significant anemia at all, others may need PRBC transfusions during the first few months of life. Splenectomy is the definitive treatment of HS; however, even in the most severe cases, this surgery is often delayed until a child is at least 5 years old given the increased risk of life-threatening sepsis with encapsulated organisms in young children.
Hereditary Elliptocytosis: The HE syndromes are a heterogeneous group of disorders characterized by the presence of elliptical-shape RBCs in the peripheral blood smear.
Common HE is a dominantly inherited condition characterized by many elliptocytes in the peripheral blood smear. The clinical severity of common HE is extremely variable, ranging from an incidental asymptomatic condition, most commonly observed, to mild-to-moderate hemolytic anemia. The clinical expression of hemolytic HE ranges from a moderate hemolytic disorder to a severe, near-fatal or fatal hemolytic anemia. Hereditary pyropoikilocytosis (HPP) is a severe hemolytic anemia, with red cell fragments, poikilocytes, and microspherocytes seen on peripheral blood smear. From a clinical perspective, it is difficult to distinguish severe hemolytic HE from HPP. Once regarded as a separate condition, HPP is now recognized to be a variant of the HE disorders. Spherocytic HE is a rare condition in which both ovalocytes and spherocytes are present on the blood smear. Southeast Asian ovalocytosis (SAO), also known as stomatocytic elliptocytosis, is an HE variant prevalent in the malaria-infested belt of Southeast Asia and the South Pacific, and it is characterized by rigid spoon-shaped cells that have either a longitudinal slit or a transverse ridge. The varied clinical and hematologic manifestations of HE are an expression of the numerous molecular defects that give rise to an elliptocytic-shape erythrocyte. The most commonly encountered cases seen are nonhemolytic common HE, hemolytic HE, and HPP.
The incidence of HE is estimated at 1:2000–4000 in the United States.27 It is found worldwide but is more common among people of African and Mediterranean decent. HE is inherited in an AD fashion. Heterozygous patients generally have asymptomatic elliptocytosis (common HE) that is often found incidentally. Overall, only 12%–15% of patients with HE are symptomatic at some time during their life.27
Similar to HS, this membrane disorder is due to qualitative and quantitative defects of RBC membrane cytoskeletal proteins. Alternation in the amount, function, and structure of these proteins leads to elliptocyte formation, instability of the RBC membrane, and in some cases hemolysis. In the last subset of HE patients, symptoms include anemia, splenomegaly, and intermittent jaundice.
Most neonates with HE are asymptomatic. However, infants with rare HE subtypes (common HE with infantile poikilocytosis and HPP) may present with severe hemolytic anemia. These neonates present with jaundice, moderate-to-severe anemia, and peripheral blood smear findings of marked poikilocytosis. In the neonatal period, common HE with infantile poikilocytosis and HPP are clinically indistinguishable. However, the poikilocytosis and severe hemolytic anemia are transient in patients with common HE. The distorted RBCs noted at birth morph into the typical HE elliptical shape by a few months of life, and there is complete resolution of the hemolytic anemia. At birth, it is difficult to predict if an infant has common HE or HPP. Hydrops fetalis has been described in association with unusually severe hemolytic HE.
Children with common HE have anywhere from a few up to all elliptocytic RBCs on their peripheral blood smear. However, these elliptocytes usually do not appear until 4–6 months of age, so infants who present to the neonatal units generally have severe hemolytic anemia with poikilocytosis. In all cases, however, infants with hemolytic HE need to be followed to see if their elliptocytosis persists or benign common HE evolves. In the hemolytic variants seen in infants, the peripheral smear may demonstrate red cell fragments, microspherocytes, and some elliptocytes. Osmotic fragility testing in these patients is abnormal, with increased fragility. In addition to typical features of hemolytic anemia, the MCV of those with HPP can be very low (25–75 fL) due to the presence of a large number of microspherocytes.
The management of neonates with hemolytic anemia due to HE is identical to any patient with hemolytic anemia. Most neonates require no treatment, but some may have significant hyperbilirubinemia and anemia requiring phototherapy, exchange transfusion, or PRBC transfusion. Splenectomy later in childhood is helpful in minimizing or resolving the chronic hemolytic anemia in those with severe hemolytic HE or HPP.
Hereditary Stomatocytoses (HSt): HSt syndromes are a group of inherited disorders characterized by erythrocytes with a mouth-shaped (stoma) area of central pallor on peripheral blood smear. HSt is associated with abnormalities in red cell cation permeability that lead to changes in red cell volume. MCV may be increased (hydrocytosis), decreased (xerocytosis), or in some cases, near normal.28 Many patients present with hemolytic anemia in the neonatal period. Pallor, jaundice, hepatosplenomegaly, and signs of gallstone disease (in older patients) are the most common physical findings. Peripheral blood smears show an increased number of stomatocytes (up to 3% is normal). The reticulocyte count is elevated during hemolysis, and the stomatocytes are osmotically fragile in the overhydrated form of stomatocytosis and are resistant to osmotic lysis in the dehydrated form. Neonates with HSt may require phototherapy and occasionally exchange transfusions. Patients with HSt do not generally require splenectomy as the results vary. There is an increased risk of life-threatening thrombosis after splenectomy.29 Cholecystectomy may be considered in patients with cholelithiasis.
Erythrocyte Hemoglobin Abnormalities
Hemoglobin is a tetrameric protein made up of 4 globin chains, each associated with a heme group. The production of globin chains is directed by the alpha (α) gene cluster on chromosome 16 and the beta (β) gene cluster on chromosome 11. The α-gene cluster contains the embryonic delta (ζ) gene and 2 adult α genes. The β-gene cluster contains the embryonic epsilon (ε) gene, the fetal gamma (γ) gene, and the adult β gene. During fetal development, embryonic globin production is switched to fetal or adult globin production at different times (ζ → α, ε → γ, and γ → β). Various globin chains form different hemoglobin tetramers during embryonic, fetal, and postnatal life (Figure 31-1). Embryonic hemoglobins (Hb Gower 1, Hb Gower 2) are responsible for oxygen delivery during the first 8 weeks of gestation. By 10 weeks, embryonic chain production ceases, γ-chain synthesis ensues, and fetal hemoglobin (HbF: α2γ2) becomes the dominant hemoglobin. HbF has a high oxygen affinity, allowing the fetus to extract oxygen across the placenta. At term, 60%–90% of hemoglobin in a newborn is HbF. Following birth, γ-chain production is replaced by β-chain synthesis. By 6 months of age, 95% of hemoglobin will be adult hemoglobin (HbA), which is composed of 2 α and 2 β chains (α2β2). At birth, production of δ chains begins. The presence of HbA2(α2δ2), a minor hemoglobin, increases gradually to the adult level of 2%–3% during the first few months of life.
Congenital hemolytic anemias can be secondary to deficient production of normal hemoglobin (thalassemia syndromes) or production of abnormal hemoglobins (sickle cell disease). The α-globin chain disorders are manifest at birth, while β-globin chain abnormalities may not be clinically apparent until 4–6 months of age, after the switch from γ- to β-globin chain synthesis. Rare mutations affecting γ-globin chains also exist, and these can result in transient neonatal problems that resolve by 3 months of age.
Thalassemia Syndromes: Thalassemia disorders are due to decreased production of normal α- or β-globin chains, leading to a relative excess of the complementary unaffected globin chains. Thalassemias are one of the most common genetic disorders worldwide. Just like G-6-PD deficiency and sickle cell anemia, it is believed that thalassemia heterozygosity is protective against malaria. In the United States, the large influx of Southeast Asian people has led to a significant increase in the number of thalassemia syndromes seen in this country. Newborn screening programs for sickle cell disease also have led to discovery of nonsickling hemoglobinopathies, including thalassemia.
α-Thalassemia Disorders: These conditions are due to decreased or absent α-globin synthesis, a consequence of deletions or mutations of α-globin genes. More than 40 different deletions have been identified.30 Most commonly, α thalassemia occurs in China and Southeast Asia and less frequently in India, Kuwait, the Middle East, Greece, Italy, and northern Europe. In African Americans, α-thalassemia trait is common, with up to 5% prevalence, but it rarely causes clinically significant problems. With increasing immigration to the United States from countries with high carrier rates of thalassemia, the prevalence of significant α thalassemia is rising. Data from the California newborn program, a state with a 13% Asian population, suggest that the prevalence rate of α-thalassemia disorder is 1/9000.31
Decreased α-globin chain production is associated with accumulation of the non-α-globin chains, leading to the formation of γ4 tetramers (Hb Bart) and β4 tetramers (HbH). The amount of Hb Bart or HbH will vary, depending on the number of α-globin genes deleted and the age of the patient.
The silent carrier state is due to deletion of one α-globin gene. There are no clinical symptoms, and the blood counts are normal.
The α-thalassemia trait is due to deletions of 2 α-globin genes. These individuals have no clinical symptoms but will have microcytosis, hypochromia, and mild anemia with a normal hemoglobin electrophoresis. At birth, the MCV is less than 100 fL, and the Hb Bart level is 2%–10% (normal is 1%). Beyond infancy, Hb Bart disappears, and the only abnormality is mild microcytosis. This is a clinically benign condition; therefore, establishing the diagnoses is not necessary in the neonatal period. In people of African extraction, α-thalassemia trait is due to 2 deletions occurring in trans (2 different chromosomes) vs cis deletions (occurring on the same chromosome), which is what is observed in Asians. If an α-thalassemia syndrome is noted in any Asian child, the parents should be referred for α-thalassemia testing and genetic counseling to determine the risk of their future pregnancies being affected by a more severe type of α thalassemia (see further discussion this chapter). The risk of more serious α-thalassemia conditions is not an issue in people of African background unless the mating partner has α-thalassemia trait and is Asian.
Hemoglobin H disease is due to deletion of 3 α-globin genes (- -/- α). Also, a combination of 2 α-globin deletions and an α-globin gene mutation, most common constant spring (- -/ααcs), results in HbH disease. The clinical features of HbH disease beyond infancy are characterized by mild-to-moderate chronic microcytic anemia with variable degrees of intermittent hemolysis. HbH is unstable and undergoes denaturation in the presence of oxidant stress, thereby forming intracellular inclusions that damage red cells, leading to early removal from circulation. Similar to G-6-PD deficiency, exposure to oxidants can lead to increased hemolysis and worsening anemia. In most cases, HbH disease does not cause clinical problems other than microcytic anemia in neonates. Diagnosis in the neonatal period generally results from newborn screening tests, which detect increased amounts of Hb Bart (usually greater than 20%). Beyond infancy, children with HbH disease have a mild-to-moderate microcytic anemia (Hb 9–10 g/dL), and there can be evidence of hemolysis, such as indirect hyperbilirubinemia and splenomegaly. These infants and children rarely require RBC transfusions.
Hb Barts hydrops fetalis is due to deletion of all 4 α-globin genes (- -/- -). There are no functional α-globin chains produced; non-α-globin chains accumulate, forming exclusively Hb Bart (γ4) and HbH (β4). Embryos that lack 4 α-globin genes are well until 8 weeks of age, when production of embryonic hemoglobin ceases. Subsequently, due to lack of α-globin chains, Hb Bart becomes the predominant hemoglobin. Hb Bart has a very high oxygen affinity and is unable to efficiently deliver oxygen. Consequently, these fetuses develop anemia and have severe hypoxia, resulting in hydrops with heart failure, generalized edema, and severe growth failure. Most homozygous α-thalassemia fetuses die in utero, although some survive until term due to persistent expression of embryonic globin chains. These hydropic infants often have congenital anomalies and significant neurologic defects. There are case reports of infants with 4 α-globin gene deletions supported with intrauterine transfusions until delivery. Postdelivery, these infants received PRBC transfusions until bone marrow transplantation. This management strategy is controversial as there remain concerns regarding poor in utero neurodevelopment and long-term outcome.
Besides the fetal effects, there are serious maternal complications in pregnancies of homozygous α thalassemia. Up to 30% of women develop severe preeclampsia. Antepartum hemorrhage, hypertension, renal failure, congestive heart failure, and placental abruption are other known complications. Without proper medical care, up to a 50% mortality rate has been reported.30 Hemoglobin Bart hydrops fetalis can be diagnosed prenatally by DNA-based testing of amniocytes from amniocentesis or chorionic villus sampling. It is important to establish this diagnosis early to avoid the serious maternal complications seen in these pregnancies.
β-Thalassemia Major: β-Thalassemia major, due to mutations in both β-globin genes, is not a neonatal disease. Anemia from β thalassemia is not notable until 3 months of age when γ-globin production decreases. There are 2 groups of β thalassemia. β0 thalassemia is caused by complete absence of β-chain production; β+ thalassemia has significantly decreased but some β-globin production. Hemoglobin electrophoresis during newborn screening of neonates with β0 thalassemia reveals HbF only, consistent with absent HbA production, while β+-thalassemia patients will have HbF and very small traces of HbA. The diagnosis of β+ thalassemia is therefore only possible after the neonatal period. Infants with β-thalassemia major will become anemic and transfusion dependent by 6–9 months of age. These patients require lifelong transfusions and treatment of iron overload. Bone marrow transplantation is the only curative option.
Hemoglobin E/β thalassemia is a result of coinheritance of HbE trait and β-thalassemia trait. HbE is a β-globin variant most commonly seen in individuals in India and Southeast Asia, including Thailand, Laos, and Cambodia. There is significant clinical heterogeneity among patients with HbE/β thalassemia, ranging from a mild hemolytic process to a severe chronic transfusion-dependent anemia, identical to that seen with β0-thalassemia major. HbE/β thalassemia is diagnosed by newborn screening hemoglobin electrophoresis, which will demonstrate an FE or FEA pattern. Just like β-thalassemia major, clinical symptoms do not arise until after 3 months of age. However, once the newborn screening tests reveal this hemoglobin disorder, the infant and family should be referred to a hematology treatment center for advice and guidance.
Sickle Cell Disease: Hemoglobin S is one of the most common β-globin mutations encountered worldwide. The most severe form of sickle cell disease results from inheritance of 2 βs mutations. This genetic pattern results in what is classically known as sickle cell anemia (HbSS disease). Inheritance of one βs mutation and one β-thalassemia trait mutation causes sickle β thalassemia (HbS-β thal), which is identical to sickle cell anemia phenotypically except there is also microcytosis. Coinheritance of βs mutations and βc mutations results in HbS-C disease, which is a milder form of sickle cell disease. All 50 US states have newborn screening programs for sickle cell disease. Hemoglobin patterns are reported in the order of decreasing hemoglobin concentration. The normal neonatal pattern is fetal/adult (FA). Infants with sickle cell anemia and HbS-β thal will have an FS pattern on their newborn screen, while HbS-C disease will provide an FSC pattern. FSA is seen in Hb S-β thal while FAS indicates sickle cell trait.
Neonates are protected from clinical symptoms of sickle cell disease due to the presence of fetal hemoglobin. However, as HbF concentration decreases after birth, and as HbS production increases, significant complications from cells sickling arise. Newborn screening for sickle cell disease was adopted to ensure early diagnosis to prevent life-threatening complications. Two major complications during infancy are sepsis from encapsulated organisms and splenic sequestration. All neonates with sickle cell anemia should be referred to a sickle cell treatment center for family education and care. Penicillin prophylaxis should begin by 2 months of age, and it is particularly important for sickle cell patients to receive all scheduled vaccines during their first year of life. Sickle cell center staff or primary physicians should provide appropriate education to parents to help them identify splenic sequestration and other sickle cell complications. Couples with a child with sickle cell disease or trait should be referred for further testing and genetic counseling.
Hypoproliferative Disorders
Hypoproliferative anemia refers to anemia caused by impaired erythrocyte production (Table 31-2) and is the least-common etiology of anemia in the neonatal period. Lack of specific growth factors stimulating erythropoiesis can lead to hypoproliferative anemia, such as the anemia of prematurity. Decreased erythrocyte progenitors, as seen in isoimmunization with Kell antibodies, can cause decreased red cell production. Abnormalities resulting in increased Epo resistance or abnormalities in Epo receptor expression can result in a decreased red cell mass. Finally, lack of specific substrates or their carriers (such as iron, folate, vitamin B12, or transcobalamin II deficiency) can also lead to deficient production of red cells.
Table 31-2 Syndromes Associated With Hypoproliferative Anemia in the Neonatal Period
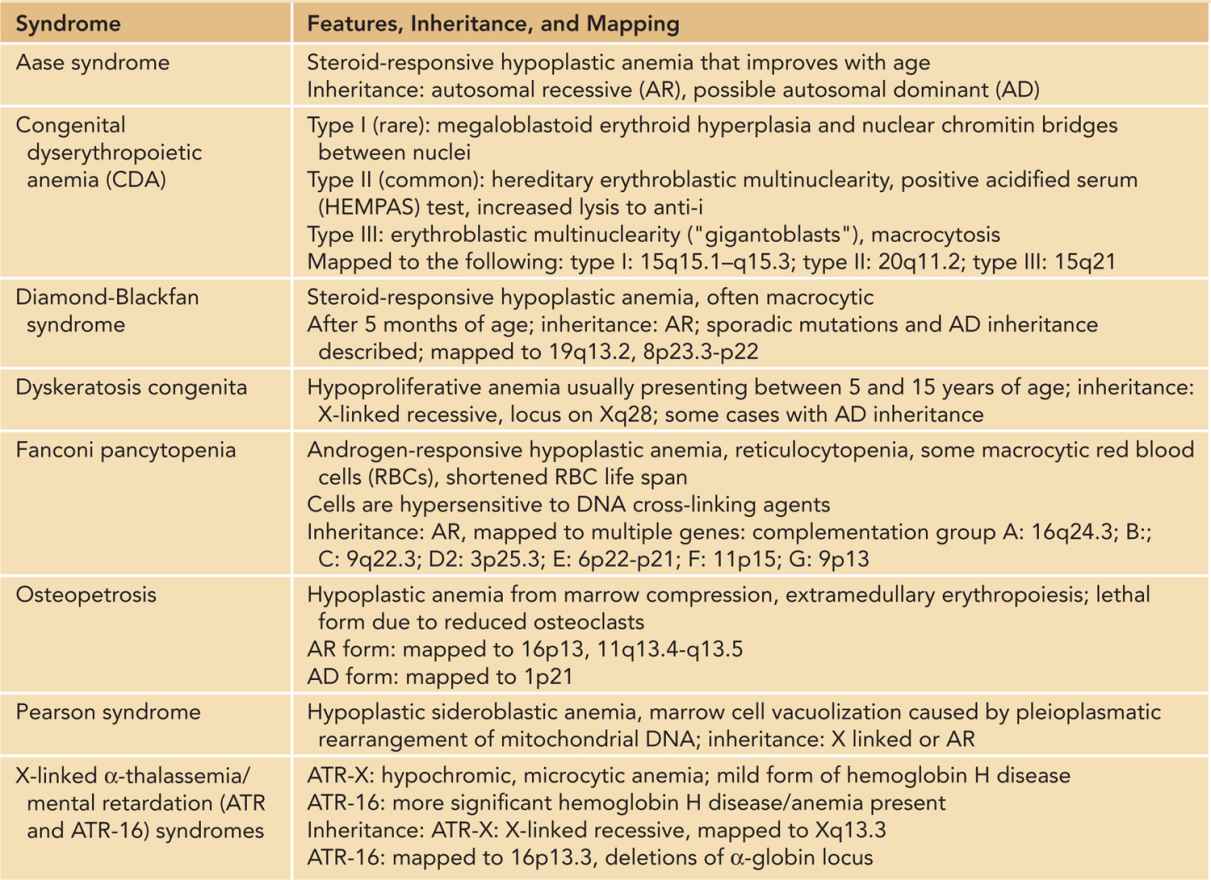
Congenital/Genetic Hypoproliferative Disorders: Congenital Hypoplastic Anemias
Congenital hypoplastic anemias refer to a rare group of inherited disorders with impaired RBC production. The most common congenital hypoplastic anemia is Diamond-Blackfan anemia (DBA). Other very rare causes include congenital dyserythropoietic anemia (CDA), and Pearson syndrome.
Diamond-Blackfan anemia is an AD disorder, estimated at 4–7 cases per million live births, occurring equally in males and females. With the discovery of multiple ribosomal gene mutations and deletions in patients with DBA, it is now firmly established that DBA is a ribosomopathy.32 Mutations in 9 different ribosomal protein genes (RPL 5, RPL 11, RPL 35A, RPS 7, RPS 10, RPS 17, RPS 19, RPS 24, and RPS 26) have been confirmed to be associated with 50% of DBA patients. An additional 20% of patients have deletions of these same ribosomal protein genes, while 30% of patients with DBA remain genetically unclassified.
In children 6 months of age to toddler years, the main differential diagnosis of RBC aplasia is DBA vs transient erythroblastopenia of childhood (TEC). However, TEC is rarely ever seen in newborns and is unusual in the first 6 months of life. Other causes of red cell aplasia include congenital parvovirus infection, maternal drug exposure, or a marrow infiltrative process such as congenital leukemia. Anemia with reticulocytopenia can also be seen in newborns who have received in utero PRBC transfusions.
Diamond-Blackfan anemia usually presents in infancy, with approximately 90% of patients diagnosed by 12 months of age. Over 25% of affected infants have severe anemia. Newborns with DBA are otherwise healthy (aside from associated congenital defects) but have severe anemia and reticulocytopenia. DBA is usually an isolated anemia, with other cell lines normal. The anemia is generally macrocytic, but the increased MCV is only appreciated beyond the neonatal period given the large RBC size during the first few months of life. If a bone marrow aspirate is done, it typically reveals normal overall cellularity with a marked decrease in erythroid precursors.
An important clinical feature of DBA is the presence of congenital anomalies in over 50% of patients. Craniofacial anomalies are most common, but upper limb, hand (particularly thumb), genitourinary, and cardiac anomalies are common. These features can be subtle and may be hard to recognize at birth. The constellation of congenital hypoplastic anemia associated with skeletal anomalies, cleft palate, growth failure, and triphalangial thumbs was previously coined Aase syndrome.33 It is now recognized that this is one of the variants of DBA. Diagnosis of DBA is supported by detecting elevated erythrocyte adenosine deaminase (eADA) activity, found in over 80% of DBA patients.34 Genetic testing for ribosomal protein mutations is available through some commercial gene diagnostic laboratories.
Packed RBC transfusions are recommended as initial treatment if needed until 1 year of age.35 After this period, steroids are started if the child remains significantly anemic. About 80% of children will respond to steroids, with reticulocytosis noted within 1–2 weeks after initiation of therapy. The natural history of DBA is variable, ranging from a need for chronic red cell transfusions, to a need for continuous administration of small doses of steroids, to spontaneous remission after years of transfusion or steroids. Treatment for those who are steroid refractory or steroid intolerant includes regular PRBC transfusions and iron chelation therapy for preventing and treating iron overload. Hematopoietic stem cell transplantation is the only curative option for DBA, and outcome of matched sibling transplants has been encouraging, with overall survival of greater than 75%. Increasing amounts of data have arisen to support the observation that DBA is a cancer predisposition syndrome. Leukemia and solid tumors have both been reported.36
Congenital dyserythropoietic anemia is a rare disorder marked by ineffective erythropoiesis, macrocytic or normocytic anemia, and characteristic abnormalities of the nuclear membrane and cytoplasm seen on electron microscopy. Both AD and AR inheritance patterns have been reported. Three types of CDA have been described.37 Type I CDA is characterized by AD inheritance, macrocytic anemia, nuclear chromatin bridges between marrow erythroblasts, and erythroid hyperplasia. Type I CDA has been linked to chromosome 15q15.1–15.3 in a large cohort, and the mutated gene is CDAN1 (codanin-1). Type II, the most common form of CDA, is characterized by normocytic anemia, erythroblastic multinuclearity, and a positive acidified serum test result. A specific mutation in the SEC23B gene on chromosome 20p has been demonstrated in this condition. CDA II appears to result from enzymatic defects in the cellular glycosylation pathway that affects band 3 anion transport activity. Type III CDA is marked by erythroblastic multinuclearity and macrocytosis.
Congenital dyserythropoietic anemia can present in the newborn period with macrocytic anemia, early jaundice, hepatosplenomegaly, and intrauterine growth retardation. Infants with type I CDA may have bony abnormalities and syndactyly. Fetuses with CDA can present with hydrops fetalis. Although rare, this disorder should be included in the differential diagnosis of newborns with anemia, jaundice, and hepatosplenomegaly. Treatment of this disorder consists of supportive therapy and close observation for side effects of chronic transfusions. Splenectomy may be helpful in some older patients with severe anemia.
Pearson marrow pancreas syndrome is a disorder involving the hematopoietic system, exocrine pancreas, liver, and kidneys. It presents in infancy with macrocytic anemia, sometimes associated with neutropenia and thrombocytopenia. The bone marrow has normal cellularity but with erythrocyte abnormalities, including vacuolization of erythroid and myeloid precursors and ringed sideroblasts. The disorder is associated with deletions in mitochondrial DNA and appears to involve defects in oxidative phosphorylation. There is no specific therapy. The disorder is usually considered fatal. Some of the few children who survive the hematologic cytopenias later go on to have the clinical and laboratory features associated with Kearns-Sayre syndrome.
Fanconi anemia is an AR-inherited disorder characterized by marrow failure. In many patients, there are congenital anomalies, including abnormalities in skin pigmentation, gastrointestinal anomalies, renal defects, and upper limb anomalies. In addition, FA is associated with an increased risk of myeloid leukemia and squamous cell cancers, occurring at a relatively younger age than when these tumors usually appear. This disorder is due to a defect in DNA repair, and 13 different FA genes have been identified.38 Peripheral blood lymphocytes are hypersensitive to DNA cross-linking agents such as diepoxybutane (DEB) and mitomycin C, representing a sensitive and specific diagnostic test for FA. The mean age of diagnosis is 6.5 years. The hematologic features of FA are rarely ever present in the newborn period. When FA is identified in neonates, it is because early recognition of congenital abnormalities initially led to specific testing for FA. Definitive treatment of FA necessitates stem cell transplantation, which can ameliorate the hematologic problems, although the increased risk of solid tumors remains.
Anemia of Prematurity
In utero, serum Epo concentrations gradually increase through the third trimester and are highest at term. Epo measurements made on neonatal cord blood of laboring and nonlaboring mothers can reflect hypoxic stress during labor and delivery. An increase in NRBCs can be seen with chronic in utero hypoxic stress; however, acute stress (less than 24 hours) may be associated with increased Epo concentrations alone. Serum Epo concentrations at birth normally range from 5 to 100 mU/mL, while Epo concentrations in anemic, nonuremic adults range from 300 to 400 mU/mL.
In preterm infants, adaptive mechanisms to the extrauterine environment are incomplete, and the transition to term organ function often follows gestational age, regardless of premature delivery. In anemic preterm infants, Epo concentrations are significantly lower than those found in adults, given the degree of their anemia. This normocytic, normochromic anemia is termed the anemia of prematurity. It commonly affects infants 32 weeks’ gestation or less and is the most common anemia seen in the neonatal period.39 The anemia of prematurity is minimally responsive to the addition of iron, folate, vitamin E, or other erythropoietic nutrients. Some infants may be asymptomatic despite hemoglobin concentrations below 8 g/dL, while others demonstrate signs of anemia that appear alleviated by transfusion. Signs associated with the anemia of prematurity include tachycardia, increased episodes of apnea and bradycardia, poor weight gain, tachypnea, an increased oxygen requirement, and elevated serum lactate concentrations that decrease following PRBC transfusion.
Multiple studies evaluating the use of recombinant Epo to prevent and treat the anemia of prematurity have been published to show that Epo is successful in preterm infants in stimulating erythropoiesis (described in the treatment section of this chapter). Epo recipients will maintain hematocrits 4%–6% greater than placebo recipients (Figure 31-2), total transfusions are decreased, and the number of nontransfused infants are increased.
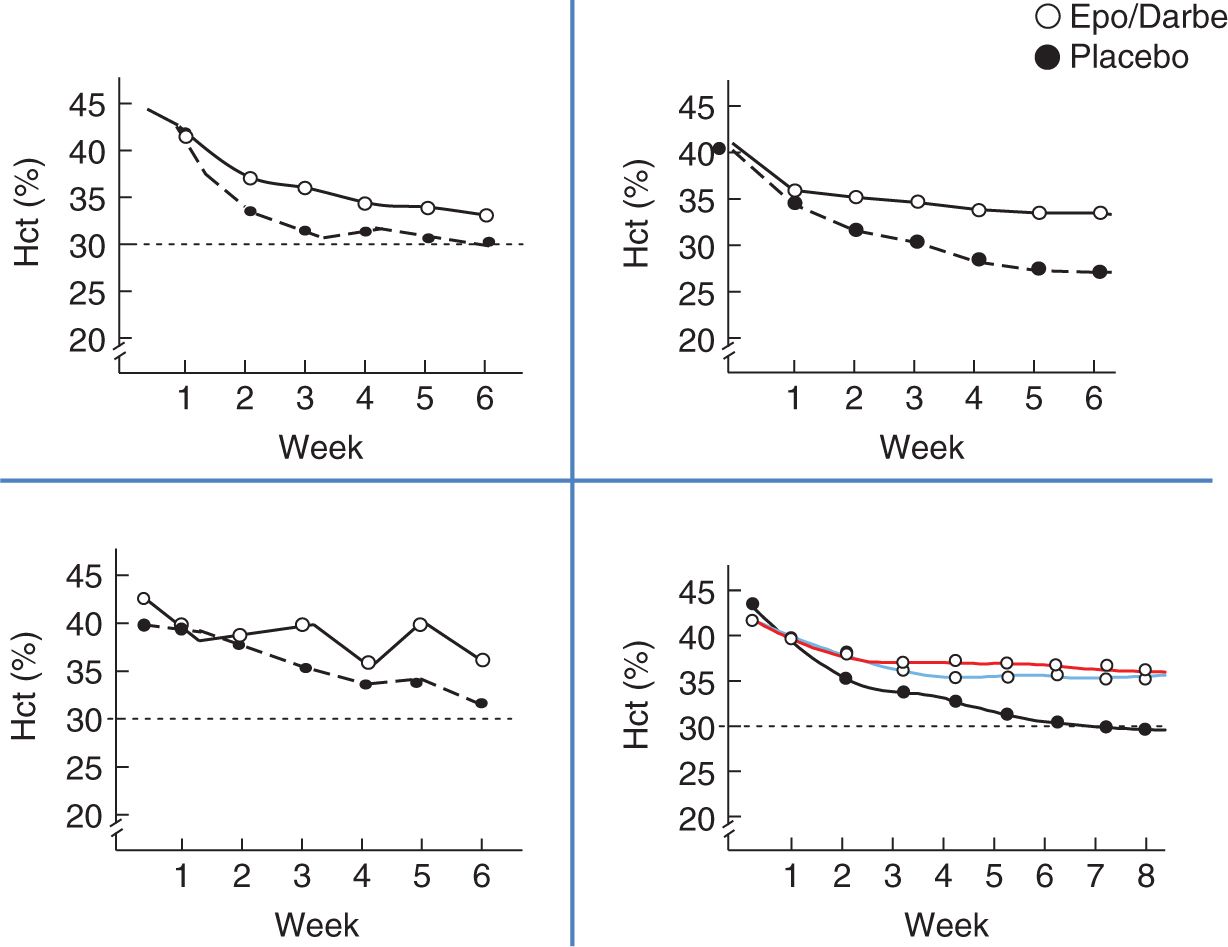
FIGURE 31-2 Hematocrit differences in erythropoietin (Epo) studies. Hct, hematocrit.57,61–63
Fetal/Neonatal Anemia due to Congenital Infection
Infections before and after birth can lead to anemia through a hemorrhagic, hemolytic, or hypoproliferative process or a combination of processes. Neonatal sepsis due to Escherichia coli, group B streptococcus, and other perinatal organisms may result in hemolysis, DIC, and hemorrhage. Infants are often jaundiced and have hepatosplenomegaly, although the degree of hyperbilirubinemia does not always correlate with the degree of anemia. Infants may have an elevated direct bilirubin as well, possibly due to infectious involvement with the liver. Bacteria such as E coli will produce hemolytic endotoxins, which result in increased red cell destruction, often associated with a microangiopathic process.
Fetal and neonatal parvovirus B19 infection can cause severe anemia, hydrops, and fetal demise.40 The infant generally presents with a hypoplastic anemia, but hemolysis can occur as well. The virus replicates in erythroid progenitor cells and shuts down erythropoiesis, resulting in aplastic anemia. In utero transfusions for hydropic fetuses have been investigated but are not successful in all patients. Treatment during aplastic crises with intravenous immunoglobulin (IVIG) leads to resolution of the anemia.
Both malaria and the human immunodeficiency virus (HIV) can be associated with neonatal anemia. Congenital malaria may occur in major urban areas, where imported cases of malaria are increasing. Congenital HIV infection can be asymptomatic in newborns. Infants born to mothers on zidovudine (AZT) may have hypoplastic anemia due to transplacental side effects of AZT.
Other Erythropoietin-Responsive Anemias in the Newborn Period
Late Anemia of Rh Disease. Early anemia in infants with Rh hemolytic disease is caused by ongoing antibody-mediated hemolysis, but a late (age 1 to 3 months) anemia resulting from diminished erythrocyte production can also occur. Late anemia appears to be more common in infants who receive intrauterine transfusions and is characterized by low serum concentrations of Epo but erythroid progenitors that remain highly responsive to recombinant Epo in vitro. Infants with this late anemia will often receive PRBC transfusions until the late anemia resolves, generally by the third or fourth month of life. Following the active hemolytic period when circulating anti-D antibody levels are elevated, the administration of Epo is effective at stimulating erythropoiesis and can serve as an alternative to erythrocyte transfusion.41 Outpatient Epo administration may also diminish or eliminate the need for hospitalization in those centers in which infants are hospitalized for transfusions.
Kell isoimmunization is a unique hemolytic disorder that selectively impacts marrow erythroid precursors, resulting in both hemolytic and hypoproliferative anemia. Kell positive infants born to Kell-negative mothers can present with severe and protracted anemia.37 Epo administration for neonatal anemia after active hemolysis is resolved can stimulate erythropoiesis.
Anemia of Bronchopulmonary Dysplasia. Anemia can develop in patients with bronchopulmonary dysplasia (BPD). The anemia of BPD is characterized as normocytic, normochromic, hyporegenerative anemia, with marrow normoblast iron stains that are distinct from those observed in both the anemia of chronic disorders and the anemia of prematurity. Similar to the late anemia of Rh hemolytic disease, the anemia of BPD responds to Epo administration. The explanation for reduced Epo production in patients with BPD requires further study to determine if other factors that could create a relatively Epo-resistant environment are involved, such as interleukin (IL) 1, tumor necrosis factor, and interferon γ.
Anemia in Neonates Requiring Surgery. Neonates who are born with problems that require surgical repair, such as congenital diaphragmatic hernia, gastroschisis, omphalocele, meningomyelocele, and craniofacial abnormalities often undergo surgery during the period of physiologic anemia. These hospitalized infants undergo blood loss through phlebotomy, the surgery itself, and postoperative care. As a result, the physiologic anemia is exacerbated, and transfusions are often administered to increase hemoglobin concentrations, especially around the time of surgery. These infants respond to Epo administration by increasing erythropoiesis42 and may also benefit from the nonhematopoietic, mitogenic effects of Epo, such as an increase in villous growth.
In addition to neonates who require surgery, other neonatal populations may benefit from Epo therapy. Epo has been used successfully to treat anemia in newborns with end-stage renal disease due to congenital nephrotic syndrome or polycystic kidney disease and in infants with congenital heart disease awaiting surgery. In addition, infants with hemolytic disease from ABO incompatibility and HS have been treated with Epo.43 Further studies are required in these populations to determine whether treatment with Epo is beneficial and does not cause further harm through increased hemolysis and hyperbilirubinemia.
Hemorrhage
The process of blood loss from the intravascular space can be either acute or chronic and can occur at any time during the prenatal, perinatal, or postnatal periods (Table 31-3). Acute hemorrhage generally results in a change in hemoglobin and hematocrit only, while chronic hemorrhage can result in changes to other red cell indices, such as MCV, MCHC, reticulocyte count, and red cell distribution width (RDW).
Table 31-3 Etiology of Neonatal/Perinatal Hemorrhage



