16.1 Anaemia
Definition
Anaemia is a common medical condition throughout all ages of childhood. However, the common causes vary with age. Anaemia refers to a reduction in haemoglobin (and hence red cell mass) below that which is considered normal for the patient in question. Normative haemoglobin data differs with age and, in teenage years, sex. Clinicians need to ensure that, when considering a diagnosis of anaemia, correct age-specific and, where applicable, sex-specific reference ranges are used. These reference ranges may vary according to the laboratory analyser in use. Thus each laboratory should report their own specific age-related reference ranges. An example of the age-related variation is shown by the reference ranges in Table 16.1.1. The majority of reference ranges in clinical use reflect 95% confidence intervals, so that 2.5% of individuals who are in fact ‘normal’ would be expected to consistently have haemoglobin levels just below the lower limit of the reported reference range.
Table 16.1.1 Normal haemoglobin values for age
| Age | Haemoglobin (g/L) |
|---|---|
| Birth | 135–200 |
| 1 month | 100–180 |
| 2 months | 90–140 |
| 6 months | 95–135 |
| 1 year | 105–135 |
| 2–6 years | 110–145 |
| 6–12 years | 115–155 |
| > 12 years (female) | 120–160 |
| > 12 years (male) | 130–180 |
Physiology
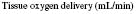
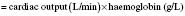
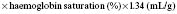
where 1.34 is a constant and represents the amount of oxygen carried by 1 g normal haemoglobin.
The key issues in this basic physiological equation are that:
• the parameters are multiplied, such that small decreases in cardiac output and haemoglobin and haemoglobin saturation lead to an overall large decrease in tissue oxygen delivery. Thus patients with cardiac disease may tolerate less reduction in haemoglobin before developing tissue hypoxia, and hence often have considerable urgency in treating their anaemia. No single haemoglobin (Hb) level can be used as an indication for transfusion therapy as these other factors need to be considered.
• in the presence of anaemia, cardiac output must be increased to maintain tissue oxygen delivery (Hb saturation cannot be increased above 100%). Failure of this compensatory mechanism or limitation of cardiac output by another disease will result in tissue hypoxia. Cardiac output is determined by cardiac stroke volume and heart rate. Therefore, heart rate is an important measure of the stress the anaemia is placing on the patient’s cardiac reserve. All anaemic patients should have their vital signs, especially heart rate and respiratory rate, assessed as part of their initial medical evaluation, and these parameters should be used to monitor progress and response to therapy.
• Hb saturation is normally close to 100% in children without cyanotic congenital heart disease or significant lung pathology. Thus, in otherwise well children with severe anaemia, or children in whom the Hb saturation is measured as 99–100%, inspired oxygen therapy makes little if any contribution to improving tissue oxygenation. Recovery of red cell mass (and hence Hb) is the most effective therapy.
• in children with cyanotic congenital heart disease or pulmonary pathology, the natural compensation for reduced Hb saturation is to increase Hb concentration. Hence, if a child with cyanotic congential heart disease who usually has a relatively increased Hb was to develop a ‘relative anaemia’, they might develop symptoms of anaemia at Hb levels that would be considered normal in most children. Treatment of ‘relative anaemia’, if required, is based on the same principles as treatment of ‘true anaemia’.
Clinical presentations
In addition, during the history and examination, other key considerations are:
• Is there evidence of cardiac decompensation or other adverse events as a result of the anaemia? This clearly makes appropriate therapy a matter of urgency.
• Are there clues to the aetiology of the anaemia?
• Is there evidence of multilineage cytopenias (neutropenia and thrombocytopenia)?
• Is there evidence of an associated, perhaps causative, disease?
Information that assists in answering these questions is shown in Table 16.1.2.
Table 16.1.2 Relevant information required on history and examination for patients with anaemia
| Critical question | Information obtained on history and examination |
|---|---|
| Cardiac decompensation | Exercise tolerance |
| Heart rate and respiratory rate | |
| Signs of congestive heart failure | |
| Altered conscious state, irritability, restlessness | |
| Aetiology | Duration of symptoms (bone marrow failure and haematinic deficiency usually have a longer duration of symptoms) |
| Family history (hereditary spherocytosis, G6PD deficiency, haemoglobinopathies and others are inherited causes of anaemia. Maternal history (e.g. veganism may be associated with B12 deficiency in infants) | |
| Birth and neonatal history (blood loss at birth, birth asphyxia and maternal blood group compatibility are all important in assessing neonatal anaemia. Jaundice at birth may give a clue to an episodic haemolytic disorder in older children) | |
| Presence or absence of jaundice (haemolysis) | |
| Drug exposure: as a cause of haemolysis, or bone marrow suppression | |
| Blood loss: trauma, recent surgery, iatrogenic in neonates, epistaxis, menstrual loss | |
| Dietary history: iron deficiency can be predicted in infants less than 12 months of age fed cow’s milk, or in toddlers who have failed to transfer to solid foods adequately | |
| Multilineage cytopenias | Bruising or bleeding, especially petechiae (thrombocytopenia) |
| Infection, mouth ulceration (neutropenia) | |
| Associated disease | Gastrointestinal symptoms (e.g. coeliac disease, inflammatory bowel disease) |
| Joint or bone pain (e.g. leukaemia, sickle cell disease, arthritis) | |
| Renal disease | |
| Malignancy | |
| Infection: as a primary cause (e.g. malaria), a precipitant of acute deterioration in a more chronic anaemia, or a trigger to acute haemolysis | |
| Neurological disorders, developmental delay/regression, failure to thrive may reflect functional B12 deficiency in infants. Pica may be associated with iron deficiency | |
| Eating disorders in older children | |
| Bleeding disorders |
Initial investigations
Examination of the blood film
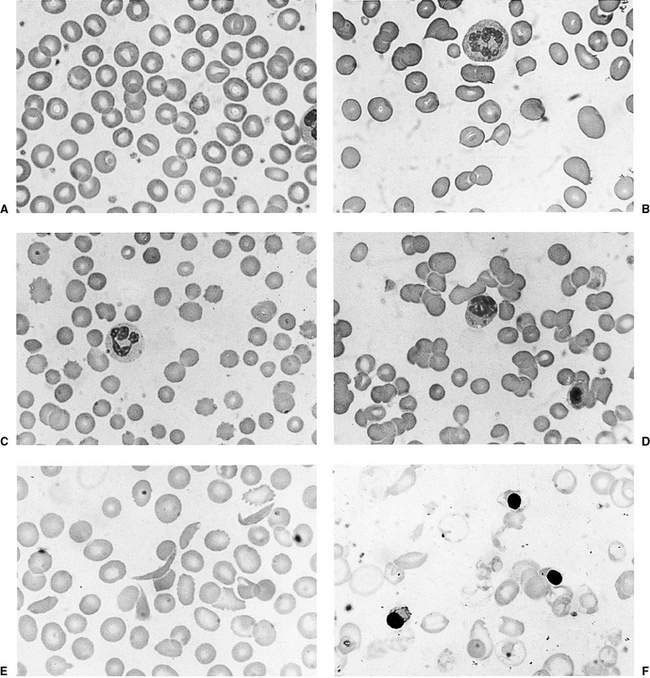
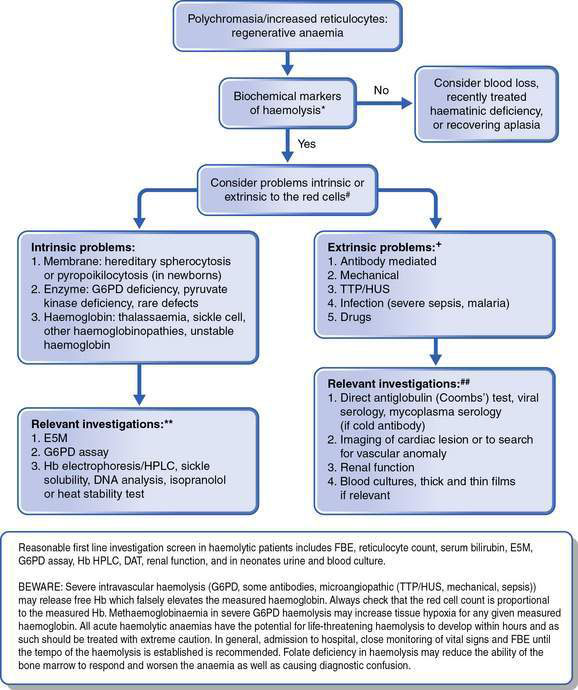
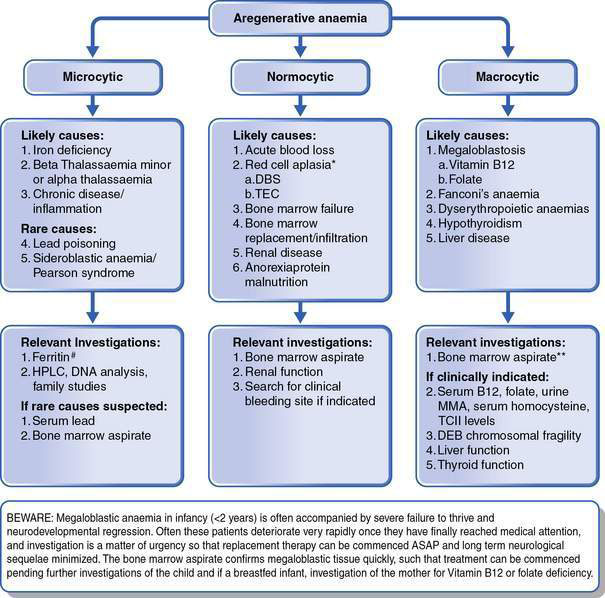
This is as important as the evaluation of the red cell indices, leukocyte count and platelets. The presence of abnormal red cell size, shape, inclusions, Hb content, and evidence of regeneration will usually suggest the cause of the anaemia and direct the next stage of investigation. The presence of abnormal leukocytes or abnormal platelet numbers may suggest a specific diagnosis such as leukaemia. Examples of a normal blood film and blood films in some conditions associated with anaemia are shown in Figure 16.1.1. Further investigations are suggested by the algorithms in Figures 16.1.2 and 16.1.3.


 Practical points
Practical pointsDetermining the urgency of investigation of anaemia
• Mild anaemia (Hb > 8 g/L) may still require urgent investigation and management, depending on the cause. Hence, until the cause of anaemia has been determined in a broad sense, discharge from emergency department/hospital should not be considered.
• Acute regenerative anaemia (blood loss or haemolysis) has the capacity rapidly to develop severe anaemia. Blood loss is usually obvious, so haemolysis must be excluded or the rate of haemolysis (multiple Hb levels over a number of hours) understood before a patient can safely leave hospital. Thus, a FBE, reticulocyte count, blood film examination and serum bilirubin are almost always indicated in initial investigations.
• Megaloblastic anaemia in infancy, irrespective of the level of anaemia, requires urgent investigation because of the potential for rapid neurological deterioration. Hence the MCV is a crucial piece of information in the initial FBE, as is the blood film examination. A history of failure to thrive and neurological impairment in infancy should lead to consideration of megaloblastosis, as anaemia is often not the presenting symptom. A FBE with careful consideration of the red cell parameters is always warranted in this circumstance.
• Anaemia as part of a multilineage failure may have a degree of urgency because of the potential for febrile neutropenia or thrombocytopenic haemorrhage.
Specific disease entities
Disorders of stem cell proliferation
Pluripotential stem cell failure (aplastic anaemia)
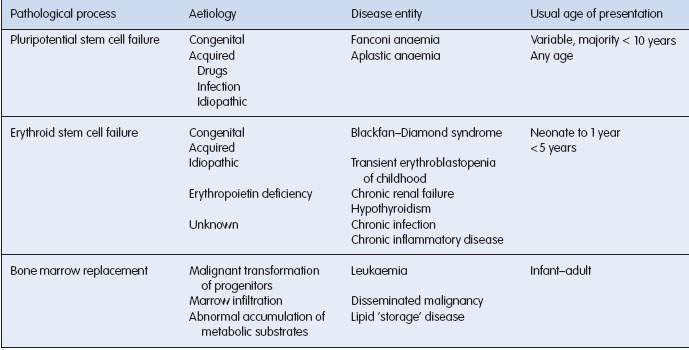
Normal marrow function is dependent on stem cell renewal and maturation of all cell lines. Failure of stem cell proliferation and differentiation results in aplastic anaemia. Both genetically determined and acquired forms occur (Table 16.1.3).
Fanconi anaemia
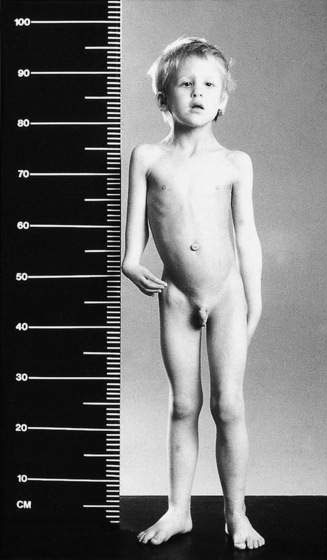
Approximately 75% of children have congenital abnormalities, with a wide range of defects. The commonest are café-au-lait spots, short stature, microcephaly and skeletal anomalies, with thumb and radial hypoplasia or aplasia being most characteristic. Renal anomalies, stenosis of auditory canals, micro-ophthalmia, hypogenitalism and a variety of anomalies of the gastrointestinal tract may also occur. The child shown in Figure 16.1.4 has many features of this disorder.
 Clinical example
Clinical exampleAcquired aplastic anaemia
• drugs: chloramphenicol, anticonvulsants, non-steroidal anti-inflammatory agents and cytotoxic drugs
• chemicals: benzene, organic solvents, insecticides
• viral hepatitis: usually non-A, non-B, non-C hepatitis, less commonly Epstein–Barr virus, cytomegalovirus, parvovirus or human immunodeficiency virus (HIV)
• preleukaemic: acute lymphoblastic leukaemia occasionally has a transient period of aplasia before the onset of the disease



