Alpha-1 Antitrypsin Deficiency Lung Disease
Terence R. Flotte and Christian Müeller
Alpha1-antitrypsin (AAT) circulates in the plasma as a 52-kD glycoprotein. It is synthesized primarily in hepatocytes and to a lesser extent in macrophages and monocytes. AAT is produced at a basal level, which results in plasma concentrations of 11 micromole or greater. AAT is also an acute phase reactant, with levels increasing dramatically during periods of stress, fever, or infection.1-6 Some individuals with AAT deficiency manifest with neonatal liver disease, as discussed in Chapter 421. This chapter discusses the pulmonary manifestations of AAT deficiency.
 EPIDEMIOLOGY AND GENETICS
EPIDEMIOLOGY AND GENETICS
Approximately 4% of northern Europeans and North Americans of European descent possess at least one mutant AAT allele. This predicts that 1 in 2500 live births in this population will possess two mutant alleles, either being homo-zygous mutant alleles or compound heterozygous mutant alleles. Up to 100,000 Americans could be AAT deficient, but only approximately 6000 Americans have been diagnosed as such. This discrepancy may represent the incomplete penetrance of the disorder or a failure to diagnose affected individuals. There is evidence that a combination of both of these explanations may be true.
Several studies have tested the utility of targeted detection of AAT deficiency within populations of adult chronic lung disease patients. These studies have consistently shown that 3% to 4% of such populations are AAT deficient. Given that approximately 11 million adults are symptomatic with chronic obstructive pulmonary disease, this targeted detection could yield a total of 30,000 to 40,000 American AAT-deficient patients. Thus, the best estimate is that there is an as-yet-undiagnosed population of 25,000 to 35,000 symptomatic AAT-deficient individuals. There may then be another 60,000 or more asymptomatic “healthy” AAT-deficient individuals.7,8
As mentioned above, mutations of the AAT gene are quite common in individuals of northern European ancestry. In particular, the one missense mutant genotype (Glu 342→Lys) that results in the PiZ phenotype on isoelectric focusing (IEF) gel electrophoresis accounts for 95% of AAT mutants in this population. Several other AAT alleles have been associated with deficiency states (eTable 516.1  ). These include numerous missense and null alleles. Environmental factors may play a role in determining which AAT-deficient individuals develop clinically evident deficiency disease. Tobacco smoke exposure (either directly from smoking or from environmental exposure) is a key risk factor for the development and severity of lung disease in AAT-deficient individuals.9 Infections have also been implicated, including both viral and bacterial infections. A number of studies have attempted to identify gene modifiers, which might significantly affect the disease phenotype in AAT-deficient individuals. Only recently has a study identified IL-10 as a potential modifier gene for the development of chronic obstructive pulmonary disease (COPD) in individuals who are PiZ homozygotes. The study suggests an association with patients carrying the high IL-10-producing allele having higher lung function; conversely, the lower IL-10-producing allele is strongly associated with lower lung function.10,11 To date, no other modifiers have yet been confirmed.
). These include numerous missense and null alleles. Environmental factors may play a role in determining which AAT-deficient individuals develop clinically evident deficiency disease. Tobacco smoke exposure (either directly from smoking or from environmental exposure) is a key risk factor for the development and severity of lung disease in AAT-deficient individuals.9 Infections have also been implicated, including both viral and bacterial infections. A number of studies have attempted to identify gene modifiers, which might significantly affect the disease phenotype in AAT-deficient individuals. Only recently has a study identified IL-10 as a potential modifier gene for the development of chronic obstructive pulmonary disease (COPD) in individuals who are PiZ homozygotes. The study suggests an association with patients carrying the high IL-10-producing allele having higher lung function; conversely, the lower IL-10-producing allele is strongly associated with lower lung function.10,11 To date, no other modifiers have yet been confirmed.
 PATHOPHYSIOLOGY
PATHOPHYSIOLOGY
Alpha1-antitrypsin (AAT), alternatively known as alpha1-proteinase inhibitor or as the Gen-Bank designation SERPINA1, is a member of the serine proteinase inhibitor (serpin) class of proteins, which have evolved over time to serve a variety of crucial roles in the maintenance of homeostasis, particularly in the context of physiological cascades such as those mediating inflammation, immunity, thrombosis, and thrombolysis. AAT itself is a serpin with multiple substrates. Its functionality in the inhibition of trypsin is the basis for its common name. Abundant evidence supports a key role in inhibition of neutrophil elastase (NE). Other neutrophil products, including proteinase 3, cathepsins, and alpha-defensins are prominent among the substrates for AAT activity.1 The various normal inhibitory capacities possessed by AAT all include regulating the inflammatory response and limiting the downstream consequences of that response. Specifically, AAT appears to limit the action of neutrophil-derived proteases to the local environment of neutro-phil accumulation, such as at a localized nidus of infection or foreign body reaction. This is particularly crucial within the lung parenchyma, where neutrophil accumulation within the pulmonary capillaries lie in close juxtaposition with interstitial elastin fibers.
Many neutrophil-derived products appear to contribute to the pathology seen in the airways and alveoli of AAT-deficient patients. These include neutrophil elastase, cathepsin G, protein-ase 3, human neutrophil, and peptide (alpha defensins). These factors, unopposed by AAT, contribute to a proinflammatory state in the airways of AAT-deficient patients and to destruction of the extracellular matrix both in the pulmonary parenchyma and in the airways. There is also evidence that such factors are injurious to the cellular components of the lung, including airway and alveolar epithelial cells and endothelial cells. 
 CLINICAL FEATURES
CLINICAL FEATURES
The majority of patients with alpha1-antitrypsin (AAT) lung disease present in adulthood. The average age of diagnosis is 52 years old. By the time of diagnosis, most patients have had pulmonary symptoms for at least 10 years. Pulmonary symptoms often include many elements in common with asthma (including bronchodilator responsive airway obstruction), emphysema, and chronic bronchitis. Patients often experience exacerbations of cough, tachypnea, and respiratory distress, which are often triggered by intercurrent illnesses, including viral respiratory tract infections.22 The radiographic appearance of the chest is quite variable but typically includes variable zones of chronic hyperinflation to a more advanced degree in the lower lobes, along with an increase in the AP diameter of the chest wall. As noted above, some individuals have coexisting lung and liver disease at the time of initial diagnosis. One or the other of these manifestations may have been underappreciated or undiagnosed prior to the definitive diagnosis of AAT deficiency. Other extrapulmonary manifestations may be seen in AAT-deficient patients, including panniculitis, which is a rare but characteristic finding.
 DIAGNOSIS
DIAGNOSIS
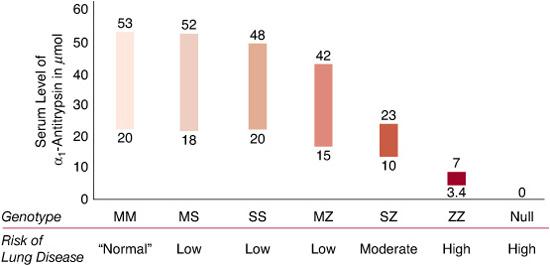
The key features for diagnosing AAT deficiency include a total plasma level less than 11 micromole (approximately 570 micrograms per mL, or 57 mg/dL) and an isoelectric focusing (IEF) gel phenotype indicating either the presence of an abnormal migration pattern or the absence of the normal M band, or both. In recent years, geno-typing technology has simplified and enhanced the ability to diagnose patients rapidly and accurately in many cases. By convention, the pheno-type as determined by IEF is designated as the Pi type, with the letters indicating all visible bands. Thus, a Z homozygote is PiZ, a normal homozygote is PiM, and a heterozygous individual is PiMZ. The corresponding genotypes would be indicated as PI*ZZ, PI*MM, and PI*MZ, respectively. The serum levels of AAT associated with various Pi phenotypes are shown in Figure 516-1. The lung pathology in AAT deficiency is classically described as panacinar emphysema, which is greater in the bases of the lung than in the apices. This type of emphysema includes loss of the entire alveolar septae and an increase in alveolar diameter. Inflammatory changes, particularly in the airways, are also typical.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


