Hypothalamo–pituitary–adrenal axis and control of cortisol secretion
Cells in the paraventricular nucleus of the hypothalamus secrete corticotrophin-releasing hormone (CRH). Released from nerve terminals in the median eminence, this 41-amino-acid peptide is transported in the hypophysial portal capillaries to the anterior pituitary corticotrophs to stimulate both the synthesis and the release of adrenocorticotrophin (ACTH).
ACTH is derived from a large precursor molecule, pro-opiomelanocortin (POMC), which is cleaved by the action of specific peptidase enzymes. The prohormone POMC can give rise to numerous hormones, including opioid peptides and melanocyte-stimulating hormone, but the main product of POMC cleavage in the corticotroph cells is ACTH. (In the brain, other products predominate.)
The main role of ACTH is to stimulate the synthesis and release of glucocorticoids and androgens from the adrenal cortex via a G-protein-coupled receptor. The immediate actions of ACTH in terms of steroid synthesis are:
increase in cholesterol esterase
transport of cholesterol into the mitochondria
binding of cholesterol to CYP11A1.
This all leads to the induction of steroidogenic enzymes and to specific structural changes such as hypervascularisation, cellular hypertrophy and cellular hypertrophy and hyperplasia.
The action of CRH on pituitary corticotrophs is potentiated by arginine vasopressin (AVP), formally known as antidiuretic hormone. AVP is secreted by parvocellular (small) neurosecretory cells into the hypophysial portal capillaries and by magnocellular (large) neurosecretory cells into the general circulation (see Chapter 16).
The production of glucocorticoids is controlled by a classic negative feedback loop in which neurons in the hypothalamus detect circulating concentrations of glucocorticoids and consequently stimulate or inhibit the release of CRH and AVP from the parvocellular neurons (Figure 17.1). The secretion of AVP by the magnocellular neurons is controlled by different stimuli, namely serum osmolality and blood volume.
In normal situations, CRH, ACTH and cortisol secretory rates demonstrate a circadian rhythm, with a peak in the early morning and a nadir in the evening. The negative feedback effect of cortisol on the anterior pituitary and the hypothalamus helps to control these increases and to regulate plasma cortisol concentrations. As ACTH also stimulates the release of androgen precursors by the adrenal gland, the secretion of these hormones parallels that of cortisol.
It should be noted that CRH is also produced in the placenta along with the binding protein CRH-BP. In this organ, CRH/CRH-BP is thought to play a role in the initiation of parturition. CRH synthesis and CRH receptors have also been identified in immune cells, where CRH has both anti‐inflammatory and pro‐inflammatory effects.
Mineralocorticoids
The secretion of aldosterone is regulated by a number of factors, the most important of which involve the renin–angiotensin system and changes in the plasma K+ concentration (Figure 17.2). Renin, secreted by the granular cells (also called juxtaglomerular cells) of the afferent arterioles at the juxtaglomerular apparatus, is an enzyme that converts angiotensinogen (produced in liver) to angiotensin 1. In the lung, angiotensin-converting enzyme (ACE) converts angiotensin I to angiotensin II, a potent vasoconstrictor and stimulator of aldosterone release by the adrenal gland.
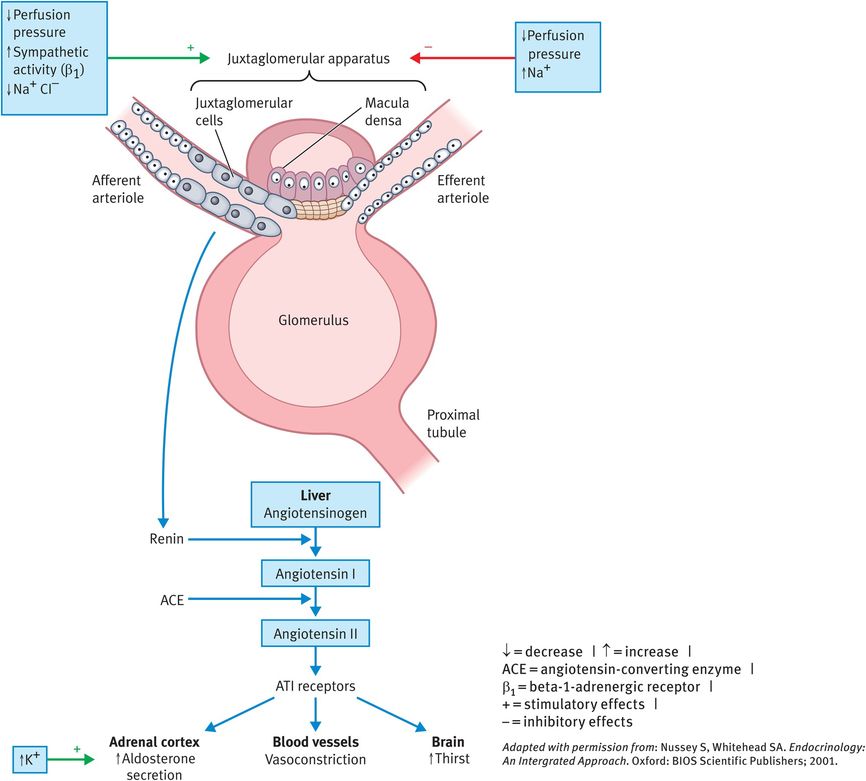
Control of aldosterone secretion
Actions of adrenocortical steroids
Glucocorticoids
Cortisol has many effects on the body:
It stimulates gluconeogenesis, the overall result being an increase in serum glucose concentrations.
It has a catabolic effect, decreasing protein stores in the body.
It has clinically significant anti-inflammatory effects (for which it is used extensively therapeutically).
It can adversely affect immunity (eosinophil and lymphocyte counts in the blood decrease with atrophy of lymphoid tissue).
Adrenal androgens
The adrenal cortex continually secretes several male sex hormones, including DHEA and its sulphate (DHEAS), androstenedione and 11-hydroxyandrostenedione, as well as small quantities of the female sex hormones progesterone and estrogen. All have weak effects, but in boys they probably have a role in the early development of the male sex organs and in girls they have an important role during puberty and menarche. Most androgenic effects result from extra-adrenal conversion of the androgens to testosterone (see Chapter 6).
Mineralocorticoids
Aldosterone accounts for 90% of mineralocorticoid activity. This hormone promotes Na+ reabsorption and K+ excretion by the renal tubular epithelial cells of the collecting and distal tubules. As Na+ is reabsorbed, H2O follows passively, leading to an increase in the extracellular fluid volume with little change in the plasma Na+ concentration. Persistently elevated extracellular fluid volumes cause hypertension. Without aldosterone, the kidney loses excessive amounts of Na+ and consequently H2O, leading to fluid depletion.
Steroid transport and metabolism
Cortisol binds to proteins in the blood, mainly cortisol-binding globulin (also called transcortin). More than 90% of cortisol is transported in the blood in this bound form.
Only 50% of aldosterone is bound to protein in the blood. All adrenocortical steroids are degraded in the liver and conjugated predominantly to glucuronides, with lower amounts of sulphates formed. About 75% of these degradation products are excreted in the urine and the rest is excreted in the stool by means of bile.
Physiology of the Adrenal Medulla
Catecholamine synthesis and secretion
The adrenal medulla constitutes less that 20% of the adrenal gland and the cells are polygonal and arranged in cords. The medulla receives blood either directly from the medullary arterioles or from the cortical venules, which are rich in cortisol and drain centripetally to the medullary venules. Epinephrine and smaller amounts of norepinephrine are synthesised by, and secreted from, the chromaffin cells of the medulla, in response to stimulation of preganglionic (cholinergic) sympathetic nerves that originate in the thoracolumbar lateral grey matter of the spinal cord. Chromaffin cells are so named because of their affinity for chromium salts, which leads to characteristic staining. As modified postganglionic nerve cells, they are classic neurosecretory cells (that is, neurons releasing hormones into the general circulation).
Like melatonin and thyroid hormones, the catecholamines are synthesised from tyrosine but, in contrast to thyroid hormones, they are made from single tyrosine molecules. These in turn are either synthesised from phenylalanine or imported from the circulation. The rate-limiting step in the synthesis of catecholamines is that catalysed by tyrosine hydroxylase, converting tyrosine to 3,4-dihydroxyphenylalanine (DOPA). The enzyme aromatic-l-amino-acid decarboxylase converts DOPA to dopamine. Hydroxylation of dopamine by dopamine β-hydroxylase leads to the formation of norepinephrine. In some adrenal medullary cells, the synthetic process stops at norepinephrine, but in most cells (particularly those at the corticomedullary junction) norepinephrine is converted to epinephrine by phenylethanolamine N-methyltransferase.
The output of the adrenal gland is controlled from nerve cells within the posterior hypothalamus, which can ultimately stimulate acetylcholine release from preganglionic nerve terminals. This induces depolarisation of the chromaffin cells and exocytosis of the catecholamine-containing granules following a transient rise in intracellular Ca2+ concentration. Once secreted, catecholamines have a very short half-life in the circulation (approximately 1–2 minutes).
Actions and metabolism of catecholamines
Catecholamines act on their target tissues through typical G-protein-linked membrane receptors. These receptors are classified as alpha-adrenergic or beta-adrenergic receptors on the basis of the physiological and pharmacological effects induced by hormone binding. The physiological effects of the catecholamines are manifold and summarised in Table 17.1. They have been characterised as preparing us for ‘fight or flight’, with overall actions comprising an increase in heart rate and stroke volume, an increase in blood pressure, a dilatation of the bronchi, the mobilisation of glucose and the stimulation of lipolysis. These actions are mediated by β-adrenergic receptors. Blood flow to the splanchnic bed is reduced by vasoconstriction of arterioles. This effect is mediated by α-adrenergic receptors and helps to divert blood flow to skeletal muscles.
| Body system | Effect | Mediator of the effect |
|---|---|---|
| Cardiovascular | Increase in heart rate and force | β1-receptors |
| Increase in venous return | α-receptors | |
| Increase in peripheral resistance | α-receptors, especially those in the subcutaneous, mucosal, splanchnic and renal vascular beds | |
| Autonomic nervous | Smooth muscle relaxation | β2-receptors |
| Smooth muscle contraction | α-receptors | |
| Modulation of fluid and electrolyte transport in the gut, kidney and gallbladder | α-receptors | |
| Fuel metabolism | Glycogenolysis, lipolysis | β-receptors |
| Increase in diet-induced and nonshivering thermogenesis | β-receptors | |
| Fluid and electrolyte balance | Decrease in Na+ excretion and glomerular filtration | Direct effects on the kidney |
| Increase in aldosterone production resulting in enhanced Na+ retention in the distal tubule | β-receptor-mediated effects on renin secretion | |
| Increase in serum K+ levels | α-receptor-mediated effects on the liver | |
| Decrease in serum K+ levels | β2-receptor-mediated effects on muscle | |
| Endocrine | Modulation of the renin–angiotensin–aldosterone system | β1-receptors |
| Increase in the secretion of glucagon and insulin | β2-receptors | |
| Inhibition of the carbachol-stimulated synthesis of catecholamines | α2-receptors |
While most catecholamines released from sympathetic nerves are taken back up into the presynaptic terminal (termed uptake 1), catecholamines released into the circulation are taken up by non-neuronal tissues (termed uptake 2) and are rapidly converted to deaminated products by monoamine oxidase (MAO) or to O-methylated products by catechol O-methyltransferase. The latter enzyme also catalyses the m–O-methylation of the products of MAO action (metanephrine, normetanephrine, epinephrine and vanillylmandelic acid). These may then be conjugated with glucuronide or sulphate and excreted in the urine.
Diseases Of The Adrenal Gland
Cushing syndrome
The term Cushing syndrome is used for an excess of glucocorticoids, however produced. The term Cushing’s disease is used when the excess of glucocorticoids is attributable to an ACTH-secreting pituitary tumour. When Cushing syndrome is caused by ectopic ACTH secretion (for example secretion by a lung tumour), the clinical features may not be typical of classic disease and include hypokalaemia and metabolic alkalosis.
The clinical features of Cushing syndrome are given in Table 17.2 and the causes in Table 17.3. When the cause is a pituitary tumour, clinical features associated with the loss of other pituitary hormones may also be apparent. These include hypothyroidism, hypogonadism and growth hormone deficiency.
| Overall frequency | Clinical feature (prevalence among those with Cushing syndrome) |
|---|---|
| Common | Moon face (with plethora) (100%) |
| Weight gain, central obesity (100%) | |
| Hypertension (80%) | |
| Mental changes (80%) | |
| Impaired glucose tolerance/diabetes mellitus (70%) | |
| Hypogonadisma (70%) | |
| Hirsutism in women (70%) | |
| Purple striae (60%) | |
| Acne (60%) | |
| Osteopenia/osteoporosis (50%) | |
| Easy bruising (50%) | |
| Proximal myopathy (50%) | |
| Less common | Poor wound healing (30%) |
| Polycythaemia (10%) | |
| Renal stones (10%) | |
| Headache (10%) | |
| Exophthalmos (10%) |
a Menstrual irregularity/infertility in women, loss of libido in men
| Overall frequency | Cause |
|---|---|
| Common (~99%) | Exogenous therapeutic glucocorticoids |
| Uncommon (<1%) | Anterior pituitary adenoma |
| Ectopic ACTH production | |
| Adrenal adenoma | |
| Rare (<0.01%) | Adrenal carcinoma |
| Ectopic CRH production | |
| Alcoholism | |
| Bilateral multinodular hyperplasia |
ACTH = adrenocorticotrophin | CRH = corticotrophin-releasing hormone
Diagnosis
Screening tests for Cushing syndrome include:
measurement of 24-hour urinary levels of free cortisol
measurement of midnight levels of salivary cortisol
low-dose dexamethasone suppression test.
The latter may be combined with the CRH test to differentiate between pseudo-Cushing caused by obesity, alcoholism or depression, pituitary Cushing due to ACTH excess and Cushing syndrome caused by primary adrenal disease or ectopic ACTH production (Table 17.4).
| Condition | Cortisol level at baseline | ACTH level at baseline | Cortisol and ACTH levels after dex administration | Cortisol and ACTH levels after CRH administration | Ratio of ACTH precursors to ACTH |
|---|---|---|---|---|---|
| Pseudo-Cushing | Normal or high | Normal or high | Both suppressed | No response | Normal |
| Pituitary Cushing | High or normal | High | Incomplete suppression | Increase by ≥40% in both | Normal |
| Adrenal adenoma | High | Normal or low | No suppression | No change | Normal |
| Ectopic ACTH secretion | High | High | No suppression | No change | High |
ACTH = adrenocorticotrophin | CRH = corticotrophin-releasing hormone | dex = dexamethasone
When investigating solid lesions of the adrenal gland, unenhanced or delayed contrast-enhanced computed tomography (CT) may help in distinguishing benign from malignant lesions by their attenuation. Benign lesions tend to have decreased attenuation because of an increased fat content. Magnetic resonance imaging (MRI) is also useful and its main benefit over CT is its improved ability, with gadolinium enhancement or with chemical shift imaging, to help in differentiating benign from malignant lesions. Ultrasonography is often the first imaging study performed in children who are suspected to have an ACTH-secreting adrenocortical tumour. It can differentiate cystic from solid adrenal masses and is useful to assess for vascular involvement and liver metastases. Radioisotope scanning is now rarely used.
For lesions of the pituitary gland, MRI is the imaging modality of choice. Visual field examination most often reveals bitemporal hemianopia if a large pituitary tumour is present.
Treatment
The definitive treatment for Cushing syndrome is surgery. This includes transphenoidal surgery for pituitary disease and adrenalectomy (either laparoscopic or conventional) for adrenal disease.
Medical management includes supportive treatment for hypokalaemia with K+-sparing diuretics and K+ supplements.
Medications used in the management of Cushing syndrome include the following:
Primary aldosteronism
An excess of mineralocorticoid steroids results in Na+ retention in the kidney with loss of K+. Na+ retention promotes H2O retention and expansion in the extracellular volume, resulting in hypertension and a suppression of renin production. This condition is also called Conn syndrome. It accounts for up to 2% of the total incidence of hypertension in the general population. The causes include:
adrenal adenoma (accounting for 80% of the total incidence)
solitary adenoma (65–70%)
multiple adenomas (13%)
microadenomatosis (6%)
adrenal gland hyperplasia (20%)
adrenal carcinoma (less than 1%).
Clinical presentation
The common clinical scenarios in which the possibility of primary aldosteronism should be considered include the following:
Patients with spontaneous or unprovoked hypokalemia, especially if the patient is also hypertensive.
Patients who develop severe and/or persistent hypokalemia in the setting of low to moderate doses of potassium-wasting diuretics.
Patients with treatment-refractory/-resistant hypertension (HTN).
Patients with severe hypokalemia report fatigue, muscle weakness, cramping, headaches and palpitations. They can also have polydipsia and polyuria from hypokalemia-induced nephrogenic diabetes insipidus. Long-standing HTN may lead to cardiac, retinal, renal and neurological problems, with all the associated symptoms and signs.
Diagnosis
The diagnosis is made from the classic biochemical findings of hypokalaemia and metabolic alkalosis associated with hyperaldosteronism.
Random plasma aldosterone/plasma renin activity (PRA) ratio is fairly constant over many physiological conditions. Simultaneous measurement of plasma renin activity (PRA) and plasma aldosterone concentration (PAC), after discontinuation of appropriate antihypertensive medication can be used in screening for hyperaldosteronism. When aldosterone is measured in ng/dL and PRA is measured in ng/mL/h, a plasma aldosterone/PRA ratio of greater than 20–25 has 95% sensitivity and 75% specificity for primary aldosteronism. When aldosterone is measured in pmol/l, a ratio greater than 900 is consistent with primary aldosteronism.
Genetic–familial primary aldosteronism
Three distinct genetic–familial varieties of primary aldosteronism exist:
type 1 variety of familial primary aldosteronism, glucocorticoid-remediable aldosteronism (GRA)
type 2 variant of familial primary aldosteronism, which is not glucocorticoid sensitive
type 3 variant of familial primary aldosteronism, which is due to potassium channel mutations.
Treatment
In 75–90% of people with a solitary aldosterone-producing tumour, surgical adrenalectomy corrects hypertension and hypokalaemia. Surgery should be avoided in bilateral hyperplasia. As a result, it is important to distinguish the two and, as with Cushing syndrome, CT or MRI scans are the best modalities to localise a tumour or establish bilateral adrenal hyperplasia. If hyperplasia is present, then pharmacotherapy is used to correct hypokalemia and treat hypertension.
Drugs that are commonly used include mineralocorticoid antagonists like spiranolactone. The drug has estrogen-like adverse effects, including impotence and gynaecomastia. Eplerenone is a selective anti-aldosterone agent, which is a specific aldosterone-receptor antagonist that does not have the additional antiandrogen effects associated with spironolactone.
GRA is treated with small doses of glucocorticosteroids (i.e., hydrocortisone, prednisone). At optimal doses, glucocorticosteroids normalise aldosterone and blood pressure.
Phaeochromocytoma
A phaeochromocytoma is a rare catecholamine-secreting tumour of the sympathetic nervous system. The majority (90%) of such tumours arise from adrenal sites, whereas 10% occur elsewhere in the sympathetic chain. Tumours that arise outside the adrenal gland are termed extra-adrenal phaeochromocytomas or paragangliomas.
Most tumours release both epinephrine and norepinephrine, but large and extra-adrenal tumours produce almost entirely norepinephrine. Excessive catecholamine secretion from phaeochromocytomas may precipitate life-threatening hypertension or cardiac arrhythmias. The tumour is malignant in 10% of cases, but may be cured completely by surgical removal. Although phaeochromocytoma has classically been associated with 3 syndromes – von Hippel-Lindau (VHL) syndrome, multiple endocrine neoplasia type 2 (MEN 2) and neurofibromatosis type 1 (NF1) – there are now 10 genes that have been identified as sites of mutations leading to phaeochromocytoma.
Diagnosis
The choice of diagnostic test should be based on the clinical suspicion of a phaeochromocytoma. Plasma metanephrine testing has the highest sensitivity (96%) for detecting a phaeochromocytoma, but it has a lower specificity (85%). In comparison, a 24-hour urinary collection for catecholamines and metanephrines has a sensitivity of 87.5% and a specificity of 99.7%. Localisation of the lesion is usually made with the help of CT or MRI scanning. A scan with iodine-123 (123I)–labelled metaiodobenzylguanidine (MIBG) is reserved for cases in which a phaeochromocytoma is confirmed biochemically but CT scanning or MRI does not show a tumour.
A somatostatin-receptor analogue, indium-111 (111In) pentetreotide, is less sensitive than MIBG. However, it may be used to visualise phaeochromocytomas that do not concentrate MIBG.
PET scanning with18F-fluorodeoxyglucose (18F-FDG), which is selectively concentrated as part of the abnormal metabolism of many neoplasms, has been demonstrated to detect occult phaeochromocytomas.
It is important to note the family history because a number of germline mutations have been identified to be responsible for familial forms of phaeochromocytoma.
Treatment
The only form of curative therapy is complete surgical removal of the tumour after initial medical treatment. Careful preoperative management is required to control blood pressure, correct fluid volume and prevent intraoperative hypertensive crises. Preoperative medical treatment reduces the risk of acute catecholamine release in response to anaesthetic drugs and surgical handling. The usual preoperative treatment is initially with α-adrenergic blockade followed by a combination of α-adrenergic and β-adrenergic blockade.
Hypoadrenalism
This condition can occur as a result of primary adrenal insufficiency (also termed Addison’s disease) but it can also be secondary to pituitary damage and results in deficient production of hormones by the adrenal cortex. The causes are summarised in Table 17.5.
| Overall frequency | Type of hypoadrenalism | Cause |
|---|---|---|
| Common (>99%) | Primary hypoadrenalism (Addison’s disease) | Abrupt cessation of exogenous sources of glucocorticoids |
| Rare (<1%) | Primary hypoadrenalism (Addison’s disease) | Autoimmunity |
| Infection (e.g. tuberculosis or fungal infections) | ||
| Haemorrhage | ||
| Metastases | ||
| Drugs (e.g. etomidate, ketoconazole, metyrapone) | ||
| Secondary hypoadrenalism | Any pituitary disease causing hypopituitarism | |
| Tertiary hypoadrenalism | Any hypothalamic disease causing hypopituitarism |
In autoimmune Addison’s disease, the autoimmune destruction of the adrenal glands may be isolated or part of multi-organ involvement. There are two types of autoimmune polyendocrinopathy. Autoimmune polyendocrinopathy syndrome (APS) type 1 is also known as autoimmune polyendocrinopathy, candidiasis and ectodermal dystrophy (APECED) syndrome. APS type 1 occurs in association with autoimmune hypoparathyroidism, chronic mucocutaneous candidiasis and other organ-specific autoimmune disorders. It is a monogenic autoimmune disorder and has autosomal recessive inheritance.
APS type 2 (Schmidt syndrome) is the association of autoimmune Addison’s disease with autoimmune thyroid disease and/or type 1 diabetes. APS type 2 has a predilection for women in middle age and the average age of onset is between 35 and 40 years. The genetic basis of isolated autoimmune Addison’s disease and APS type 2 is not clearly understood. Other autoimmune disorders can be associated with APS type 2 and these include primary gonadal failure, pernicious anaemia, vitiligo and alopecia. Antibodies to the adrenal cortex mediate autoimmune destruction of the adrenal glands.
The clinical features of Addison’s disease are given in Table 17.6. High concentrations of ACTH result in buccal pigmentation and pigmentation of scars and palmar creases.
| Overall frequency | Clinical feature (prevalence among those with Addison’s disease) |
|---|---|
| Common | Weakness (100%) |
| Weight loss (95%) | |
| Pigmentation (95%) | |
| Anorexia (95%) | |
| Nausea (95%) | |
| Less common | Abdominal pain (30%) |
| Postural hypotension (25%) | |
| Vitiligo (20%) | |
| Salt craving (15%) | |
| Aches and pains (10%) | |
| Hypoglycaemia (in adults <1%) |
Diagnosis
The diagnosis of hypoadrenalism may be suspected from evidence in routine biochemical tests of hyponatraemia, hyperkalaemia and hypercalcaemia. Biochemical confirmation may be made using a tetracosactrin test. Tetracosactrin is a peptide identical to the 24 amino-terminal amino acids of ACTH. ACTH and cortisol are measured in peripheral venous blood before a single injection of 250 micrograms of peptide and cortisol is assayed again 30 minutes thereafter. Two criteria are necessary for diagnosis: (1) an increase in the baseline cortisol value of 7 micrograms /dl or more and (2) the value must rise to 20 micrograms or more in 30 or 60 minutes, establishing normal adrenal glucocorticoid function. The absolute 30- or 60-minute cortisol value carries more significance than the incremental value, especially in patients who may be in great stress and at their maximal adrenal output. If the cortisol response is impaired and the baseline ACTH level is high, the diagnosis is primary adrenal failure. If the baseline ACTH level is low, the diagnosis is secondary adrenal failure. Stimulation of the adrenal gland in primary disease will not result in the release of cortisol. In secondary insufficiency, the atrophic adrenal glands still produce some cortisol when exposed to ACTH and the serum cortisol level rises to more than 1000 nmol/l.
Imaging studies can be useful in helping to elucidate the cause of hypoadrenalism. Both CT and MRI demonstrate a reduced adrenal gland size in people with autoimmune destruction and an enlarged adrenal gland in people with infection such as tuberculosis. MRI is superior to CT in differentiating adrenal masses but MRI cannot distinguish a tumour from an inflammatory process.
Treatment
In patients in acute adrenal crisis, intravenous access should be established urgently, and an infusion of isotonic sodium chloride solution should be begun to restore volume deficit and correct hypotension. Some patients may require glucose supplementation. The precipitating cause should be sought and corrected where possible. Treatment of acute adrenal insufficiency requires immediate intravenous administration of 100 mg hydrocortisone followed by administration of 100–200 mg hydrocortisone over 24 hours under close monitoring. Individuals with adrenal insufficiency who require long-term replacement usually receive two or three daily doses of hydrocortisone. An extra dose of hydrocortisone may be required before strenuous exercise and doses are usually doubled for intercurrent illness. The standard mineralocorticoid replacement is fludrocortisone acetate at 100–150 micrograms/days. The dose of fludrocortisone acetate is titrated individually on the basis of the clinical examination (mainly the body weight and arterial blood pressure) and the levels of plasma renin activity. As long as the patient is receiving 100 mg or more of hydrocortisone in 24 hours, no mineralocorticoid replacement is necessary. The mineralocorticoid activity of hydrocortisone in this dosage is sufficient. In women, the adrenal cortex is the primary source of androgen in the form of DHEA and DHEAS. Although the physiological role of these androgens in women has not been fully elucidated, their replacement is being increasingly considered in the treatment of adrenal insufficiency. Patient education is important for long-term care of these patients:
Patients should wear an emergency medical alert bracelet.
Patients should be instructed to double or triple their steroid replacement doses in stressful situations, such as having a common cold or tooth extraction.
Patients should be instructed to contact their regular physician or to go to the emergency department in case of illness.
Patients should be instructed on how to give themselves intramuscular injections. They should be given a prescription for parenteral hydrocortisone for use on occasions when oral intake may not be possible or when marked vomiting or diarrhoea occurs.
No adjustment needs to be made on the mineralocorticoid replacement dose in stressful situations.
Congenital adrenal hyperplasia
The term congenital adrenal hyperplasia (CAH) encompasses a group of autosomal recessive disorders, each of which involves a deficiency of an enzyme involved in the synthesis of cortisol, aldosterone or both. Deficiency of 21-hydroxylase, resulting from mutations or deletions of CYP21A, is the most common form of CAH, accounting for more than 90% of cases.
The clinical phenotype of CAH depends on the nature and severity of the enzyme deficiency. Although the presentation varies according to chromosomal sex, the sex of a neonate with CAH is often initially unclear because of genital ambiguity. If the defect is severe and results in salt wasting, then the presentation is at age 1–4 weeks with failure to thrive, recurrent vomiting, dehydration, hypotension, hyponatremia, hyperkalemia and shock (classic salt-wasting adrenal hyperplasia).
Clinical presentation
Females with severe CAH due to deficiencies of 21-hydroxylase, 11-β-hydroxylase or 3-β-hydroxysteroid dehydrogenase have ambiguous genitalia at birth (classic virilising adrenal hyperplasia).
Females with mild 21-hydroxylase deficiency are identified later in childhood because of precocious pubic hair, clitoromegaly or both, often accompanied by accelerated growth and skeletal maturation (simple virilising adrenal hyperplasia).
Females with still milder deficiencies of 21-hydroxylase or 3-β-hydroxysteroid dehydrogenase activity may present in adolescence or adulthood with oligomenorrhea, hirsutism and/or infertility (nonclassical adrenal hyperplasia).
Females with 17-hydroxylase deficiency appear phenotypically female at birth but do not develop breasts or menstruate in adolescence; they may present with hypertension.
Males with 21-hydroxylase deficiency have normal genitalia.
Diagnosis
The diagnosis of CAH depends on the demonstration of inadequate production of cortisol, aldosterone or both in the presence of accumulation of excess concentrations of precursor hormones. In 21-hydroxylase deficiency there is high serum concentration of 17-hydroxyprogesterone (usually greater than 1000 ng/dl) and urinary pregnanetriol (metabolite of 17-hydroxyprogesterone) in the presence of clinical features suggestive of the disease. In nonclassical cases, a testacosactrin-stimulation test is used to diagnose CAH. In 11-β-hydroxylase deficiency cases there are excess serum concentrations of 11-deoxycortisol and deoxycorticosterone, or an elevation in the ratio of 24-hour urinary tetrahydrocompound S (metabolite of 11-deoxycortisol) to tetrahydrocompound F (metabolite of cortisol). In addition, in the salt-wasting forms of CAH, there are low serum aldosterone concentrations, hyponatremia, hyperkalaemia and elevated plasma renin activity (PRA), indicating hypovolaemia.
Imaging studies include pelvic ultrasonography in an infant with ambiguous genitalia to demonstrate a uterus or associated renal anomalies. A bone-age study is useful in evaluating for advanced skeletal maturation in a child who develops precocious pubic hair, clitoromegaly or accelerated linear growth. Another useful test is a karyotype, to establish the chromosomal sex. Genetic testing is essential for genetic counselling and prenatal diagnosis of adrenal hyperplasia. Newborn screening programmes for 21-hydroxylase deficiency may be life-saving in an affected male infant who would otherwise be undetected until presentation with a salt-wasting crisis.
Treatment
Patients presenting with a salt‐wasting crisis are treated with intravenous sodium chloride to restore intravasular volume and blood pressure. Dextrose must be included in the rehydration fluid after the bolus dose to prevent hypoglycaemia. After samples are obtained to measure electrolyte, blood sugar, cortisol, aldosterone and 17-hydroxyprogesterone concentrations, the patient should be treated with glucocorticoids. After the patient’s condition is stabilised, all patients are treated with long-term glucocorticoid or fludrocortisone replacement (or both).
The Endocrine Society’s 2010 clinical practice guidelines note the following:
Prenatal treatment for CAH should be regarded as experimental.
Glucocorticoid therapy should be carefully titrated to avoid Cushing syndrome.
Mineralocorticoid replacement is encouraged; in infants, mineralocorticoid replacement and sodium supplementation are encouraged.
The use of agents to delay puberty and promote growth are experimental.
Psychiatric support should be encouraged for patients with adjustment problems.
Medication should be used judiciously during pregnancy and in symptomatic patients with nonclassical CAH.
Surgical care
Infants with ambiguous genitalia require surgical evaluation and plans for corrective surgery. The traditional approach to the female patient with ambiguous genitalia due to adrenal hyperplasia is clitoral recession early in life followed by vaginoplasty after puberty. Some female infants with adrenal hyperplasia have only mild virilisation and may not require corrective surgery if they receive adequate medical therapy to prevent further virilisation.
The Endocrine Society’s 2010 clinical practice guidelines note the following:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


