Acute Renal Failure
Prasad Devarajan
Acute renal failure (ARF) is classically defined as a rapid decline in glomerular filtration rate (GFR), leading to accumulation of nitrogenous wastes such as blood urea nitrogen (BUN) and creatinine. ARF is a common condition, associated with serious consequences and unsatisfactory therapeutic options.1-18 Oliguria, defined as a urine output of less than 0.5 ml/kg/hour, is an important clinical sign but occurs in only about half the cases. ARF may be classified as (1) prerenal azotemia, due to a functional response of structurally normal kidneys to hypoperfusion; (2) intrinsic ARF, due to structural damage to the kidneys from prolonged ischemia, nephrotoxins, sepsis, or intrinsic renal disease; and (3) postrenal ARF, due to obstruction of the urinary tract. 
Prerenal azotemia is usually rapidly reversed by restoration of renal perfusion, but early treatment is essential in order to prevent the progression to intrinsic ARF. Once established, there is no effective treatment for ARF, and the clinician can provide only supportive care with dialysis. While the worst outcomes are encountered in patients requiring dialysis, even mild degrees of ARF, such as occurs with only a small increases in serum creatinine, is predictive of an increase in mortality and morbidity rate, irrespective of the underlying cause.19-22
EPIDEMIOLOGY
Pediatric studies from the 1980s and 1990s report hemolytic uremic syndrome, other primary renal causes, and infections as the most prevalent causes leading to acute renal failure (ARF).23 More recent studies in developed countries show a dramatic shift in the epidemiology of ARF such that it is primarily a hospital-acquired illness, with the most common causes being renal ischemia, nephrotoxin use, congenital heart disease, bone marrow transplantation, and sepsis.24,25 In contrast, in the undeveloped world of Africa and tropical Asia, children are most likely to develop ARF secondary to gastroenteritis, septicemia, acute glomerulonephritis, or falciparum malaria. Hemolytic uremic syndrome and leptospirosis are common in Latin America, and ARF due to snakebites is often encountered in rural Asia.
Hospital-acquired pediatric ARF rates are escalating at an alarming rate, over ninefold from the 1980s through 2004.26 The incidence of the most severe forms of ARF, defined by dialysis requirement, ranges from 1% to 2% of all critically ill children.26,27 When less strict criteria for ARF are used, such as doubling of serum creatinine, the incidence rises to 21% of children in intensive care units.28 The incidence jumps to 82% in critically ill children who receive invasive mechanical ventilation and at least one vasoactive medication.28 ARF also complicates up to 24% of neonatal intensive care unit admissions and is present in 60% of neonates with severe asphyxia.29 Approximately 40% of patients with sepsis develop ARF, with 20% requiring dialysis.7
In children undergoing cardiopulmonary bypass, the incidence of ARF is in the range of 10% to 40%, depending on the definition used.30-34 In children receiving stem cell transplants, the incidence of ARF (defined by doubling of serum creatinine) is about 20%.35
PATHOPHYSIOLOGY
 PRERENAL AZOTEMIA
PRERENAL AZOTEMIA
Prerenal acute renal failure (ARF), or azotemia, is an appropriate functional response of structurally normal kidneys to hypoperfusion. The oliguria in this situation represents a renal mechanism for preserving intravascular volume. Table 471-1 lists the common causes of prerenal azotemia. A reduction in circulatory volume evokes a systemic response aimed at normalizing intravascular volume at the expense of glomerular filtration rate (GFR). Baroreceptor-mediated activation of the sympathetic nervous system and renin-angiotensin axis results in afferent renal vasoconstriction and the consequent reduction in GFR (Fig. 471-1). However, in response to renal hypoperfusion, several intrarenal autoregulatory mechanisms help maintain GFR (Fig. 471-2). Prolonged reduction in renal perfusion pressure (eg, due to dehydration or systemic hypotension) can overwhelm these compensatory mechanisms, and intrinsic ARF can ensue.
Table 471-1. Causes of Acute Renal Failure
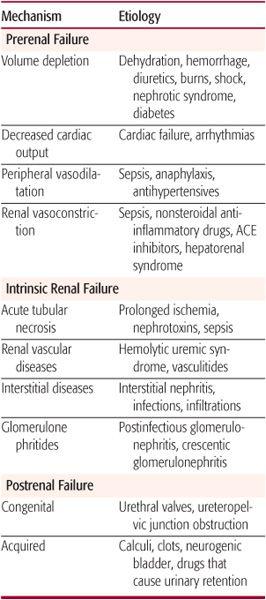
The most effective compensatory mechanism involves the intrarenal generation of vasodilatory prostaglandins that dilate the afferent arterioles.36 Nonsteroidal anti-inflammatory drugs (NSAIDs) inhibit this response and can precipitate ARF, especially in the presence of decreased circulatory volume.37 Small children who are febrile and then become dehydrated are at particular risk for developing ARF when treated with NSAIDs unless hydration is maintained when these drugs are used. A second mechanism results from a differential in the constrictor effect of angiotensin II on the efferent versus afferent arteriole. Angiotensin II constricts both the afferent and efferent arteriole, but this effect is more marked in the efferent arteriole, leading to increased hydrostatic pressure across the glomerulus and maintenance of GFR.38 Angiotensin-converting enzyme inhibitor (ACEI) therapy can interfere with this compensatory mechanism. If patients taking ACEIs become dehydrated, GFR will decline and ARF can develop. A third mechanism, termed myogenic autoregulation, refers to the unique ability of the afferent arterioles to rapidly vasodilate in response to a decrease in lateral stretch caused by hypoperfusion. Immunosuppressant medications commonly used in patients after kidney transplantation (eg, cyclosporine) can interfere with the myogenic mechanism.
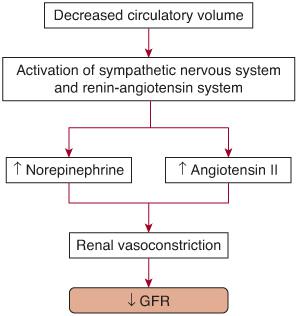
FIGURE 471-1. Pathophysiological mechanisms leading to decreased glomerular filtration rate (GFR) in prerenal acute renal failure. Baroreceptor-mediated activation of the sympathetic nervous system and renin-angiotensin axis results in afferent renal vasoconstriction and a resultant reduction in GFR.
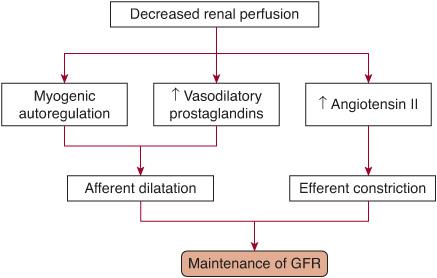
FIGURE 471-2. Renal compensatory mechanisms that maintain glomerular filtration rate (GFR) in prerenal acute renal failure. Intrarenal generation of vasodilatory prostaglandins and intrinsic myogenic mechanisms dilate the afferent arterioles. This effect combined with a differential greater constrictor effect of angiotensin on the efferent arteriole versus the afferent arteriole leads to an increased hydrostatic glomerular arteriole pressure with a resultant increase in GFR. Iatrogenic interference with these mechanisms can precipitate a reduction in GFR.
 INTRINSIC ACUTE RENAL FAILURE
INTRINSIC ACUTE RENAL FAILURE
Intrinsic acute renal failure (ARF) is most frequently caused by prolonged ischemia, nephrotoxins, or sepsis (Table 471-1) In clinical practice, it is frequently multifactorial, often occurring concomitantly, and there are overlapping pathogenetic mechanisms. Intrinsic ARF is associated pathologically with acute tubular necrosis (ATN); therefore, the terms intrinsic ARF and ATN are often used interchangeably. However, ATN is a misnomer, since frank tubular cell necrosis is rarely found in human ARF. Instead, there is effacement and loss of proximal tubule brush border, patchy loss of tubule cells, focal proximal tubular dilatation and distal tubular casts, and areas of cellular regeneration. Necrotic cell death is restricted to the highly susceptible outer medullary regions (the S3 segment of the proximal tubule and medullary thick ascending limb of Henle’s loop). More recently, apoptosis has been reported in both distal and proximal tubules, in both ischemic and nephrotoxic forms of human ARF.40,41 In addition, peritubular capillaries in the outer medulla have been shown to display a striking vascular congestion and leukocyte accumulation.42-47 The mechanisms underlying these morphological changes are detailed below.
There is evidence for hemodynamic alterations in intrinsic ARF. Total renal blood flow is reduced to about 50% of normal due to persistent intense renal vasoconstriction, as shown in Figure 471-3. Cellular mechanisms underlying these hemodynamic alterations relate primarily to endothelial damage.43-46 This leads to a local imbalance of vasoactive substances, including enhanced release of the vasoconstrictor endothelin and reduced release of vasodilatory endothelium-derived nitric oxide. However, these hemodynamic abnormalities do not account for the profound loss of renal function, and several human trials of vasodilators such as dopamine have failed to demonstrate improvement in GFR in established ARF despite augmentation of total renal blood flow.48
Proposed alterations in tubular dynamics in established ARF include obstruction, back-leak, and activation of tubuloglomerular feedback. Their interplay is illustrated in Figure 471-3. It is unlikely that obstruction alone can account for the profound dysfunction in clinical ARF, since human studies using forced diuresis with furosemide or mannitol do not show an impact on the survival and renal recovery rate of patients with established ARF.49,50 Similarly, although movement of the glomerular filtrate back into the circulation has been shown to occur, this accounts for only a very minor component of the decrease in glomerular filtration rate (GFR) in human ARF.
The role for activating tubuloglomerular feedback is controversial. 
In injured kidney tubule cells, a rapid and profound reduction in intracellular ATP content occurs, which triggers several metabolic events (see eFig. 471.1  ).
).
There is now substantial evidence for the role of reactive oxygen species in the pathogenesis of ARF.55 During reperfusion, hydrogen peroxide and superoxide are generated in tubule cells. In the presence of iron, hydrogen peroxide forms the highly reactive hydroxyl radical. Several scavengers of reactive oxygen molecules protect against ARF in animals, but human studies have been inconclusive. Two major advances have recently emerged in the area of iron chelation. The first is the availability of human apotransferrin, an iron-binding protein, which protects against renal ischemia-reperfusion injury in animals by abrogating renal superoxide formation.56 The second is the discovery of neutrophil gelatinase-associated lipocalin (NGAL), an endogenous iron-transporting protein. NGAL is one of the most highly induced genes and proteins in the kidney following early ischemic and nephrotoxic injury.57 Exogenous administration of NGAL provides significant functional protection in animal models of ARF.58 The potential use of these endogenous agents in human ARF is currently under investigation.
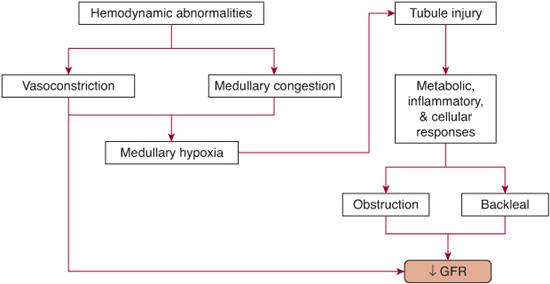
FIGURE 471-3. Pathophysiological mechanisms leading to decreased glomerular filtration rate in intrinsic acute renal failure.
The tubule cell’s biological response to acute injury is multifaceted and includes loss of cell polarity and brush borders, cell death, dedifferentiation of viable cells, proliferation, and restitution of a normal epithelium (see eFig. 471.2  ).
).
Following kidney injury, the mode of tubule cell death depends primarily on the severity of the insult and the resistance of the cell type. Necrosis occurs following more severe injury and in the more susceptible proximal tubules, whereas apoptosis predominates after less severe injury and especially in the ischemia-resistant distal nephron segments. Considerable attention has recently been directed toward unraveling the molecular pathways involved in renal tubule cell apoptosis (eFig. 471.3  ).1,11 Inhibition of apoptosis holds promise in ARF.
).1,11 Inhibition of apoptosis holds promise in ARF.
Renal tubule cells possess a remarkable ability to regenerate and proliferate after acute injury.72 Understanding the molecular mechanisms of repair may provide clues toward accelerating recovery from ARF. The potential use of progenitor cells and stem cells as therapeutic strategies in ARF is currently in the initial stages of investigation.74,75
Understanding the molecular mechanisms of repair may provide clues toward accelerating recovery from ARF. The potential use of progenitor cells and stem cells as therapeutic strategies in ARF is currently in the initial stages of investigation.74,75
Inflammation plays an important role in the pathogenesis of ARF. The major components of this response include endothelial injury, leukocyte recruitment, and production of inflammatory mediators by tubule cells (eFig. 471.4  ).
).
Strategies that modulate the inflammatory response may provide significant beneficial effects in human ARF.
 POSTRENAL ARF
POSTRENAL ARF
Postrenal ARF is a result of bilateral obstruction of the outflow tracts and is uncommon beyond the neonatal period. Postrenal azotemia is usually reversed by relief of the obstruction but is accompanied by a very significant postobstructive diuresis. The common causes of postrenal ARF are listed in Table 471-1.
CLINICAL FEATURES AND DIAGNOSTIC EVALUATION
Acute renal failure most commonly presents with a progressive accumulation of nitrogenous wastes in a predisposed patient. Less frequently, one encounters an unexplained rise in blood urea nitrogen (BUN) and creatinine. The evaluation of patients requires a complete history, physical examination, laboratory evaluation, renal imaging, and (very rarely) a kidney biopsy. It is important to recognize that early in the course of acute renal failure (ARF), changes in BUN and creatinine do not provide an accurate reflection of renal function. Despite the characteristically precipitous reduction in glomerular filtration rate that occurs in ARF, the BUN usually rises by only about 20 mg/dl per day and the serum creatinine by 1 to 2 mg/dl per day, until the values reach a steady state that more accurately represents the actual degree of renal dysfunction. This relationship is important from several perspectives: (1) it distinguishes ARF from chronic renal failure (CRF), since the BUN and creatinine in CRF do not progressively increase but display a steady high value; (2) the timing of the original insult that leads to the development of ARF can be estimated; and (3) the BUN and serum creatinine are delayed and unreliable biomarkers of ARF. In the case of BUN, an increase can be encountered in the absence of ARF, especially in patients undergoing steroid therapy, total parenteral nutrition, and bleeding within the gastrointestinal tract. For serum creatinine measurements, even normal individuals can display significant variations, depending on age, diet, medications, hydration status, muscle mass, and muscle metabolism. Furthermore, a substantial loss of GFR may occur before an increase in serum creatinine can be measured.
In patients with suspected ARF, the diagnostic evaluation is directed toward identifying the underlying cause, distinguishing between prerenal and intrinsic ARF, and discriminating between ARF and chronic renal failure (CRF).
 IDENTIFYING THE UNDERLYING CAUSE
IDENTIFYING THE UNDERLYING CAUSE
A detailed history and physical examination directed toward the salient aspects listed in Table 471-2, combined with a careful urinalysis, will yield the etiology of ARF in the majority of cases. Typically, the urine in prerenal azotemia contains only a few hyaline and fine granular casts, with little protein or blood. In contrast, proteinuria and hematuria are prominent in intrinsic ARF. Heme-positive urine in the absence of RBCs in the sediment suggests hemolysis or rhabdomyolysis. Urine microscopy reveals dysmorphic RBCs in primary glomerular disease and reveals WBCs in pyelonephritis or interstitial nephritis. Broad brown granular casts are typically encountered in ischemic or toxic ATN, whereas RBC casts are characteristic of acute glomerulonephritis, and WBC casts indicate interstitial nephritis or pyelonephritis.
 DISTINGUISHING BETWEEN PRERENAL AND INTRINSIC ARF
DISTINGUISHING BETWEEN PRERENAL AND INTRINSIC ARF
This distinction is based on the principle that prerenal azotemia is associated with maximal reabsorption of solutes and water by the intact proximal tubule, whereas the tubule cell damage typical of intrinsic ARF results in impaired reabsorptive capacity of the proximal tubule. Urinary indices based on this principle are shown in Table 471-3. The fractional excretion of sodium (FENa) is reportedly the most accurate in making this distinction. It is calculated from measured concentrations of sodium (Na) and creatinine (Cr) in the urine (U) and plasma (P), as follows:
Table 471-2. Evaluation of Acute Renal Failure



