Acute Metabolic Dysfunction
Ralph J. DeBerardinis
Inborn errors of metabolism are genetic diseases caused by mutations in metabolic enzymes, nutrient transporters, and related genes. Because metabolism is the foundation of basic processes such as energy homeostasis, fuel storage, and growth, it is not surprising that many inborn errors of metabolism present with severe, multisystem failure culminating in shock, cardiorespiratory collapse, and coma. Therefore, inborn error of metabolism is a crucial category of diseases in pediatric emergency medicine and critical care. Their pathophysiology involves a combination of disturbances emanating from the dysfunctional metabolic pathway, including failure to produce energy and required metabolites, accumulation of toxic intermediates, and complex effects on global metabolism due to sequestration of cofactors.
Inborn errors of metabolism are by definition chronic diseases, which may present with acute, life-threatening metabolic dysfunction1 (Table 114-1). Many of these “decompensating” metabolic conditions can be stabilized with rather straightforward interventions if the clinician recognizes the presence of an inborn error of metabolism and takes appropriate action. The difficulty and danger in decompen-sated inborn errors of metabolism is that their relatively low incidence and nonspecific symptoms at presentation may not trigger suspicion of a metabolic disorder. However, a few simple analytical considerations will prompt the astute clinician to recognize an inborn error of metabolism in an acutely ill child. First, such children often have clues within their clinical presentations that can be elicited by asking questions like the ones in Table 114-2. If these questions suggest the possibility of an inborn error of metabolism, the clinician should ensure that several metabolic screening laboratory studies (Table 114-3) are performed to add supporting clinical information and to guide presumptive management and specialized diagnostic testing.
Regardless of a patient’s clinical history, inborn error of metabolism should be considered in all newborn babies with serious unexplained illnesses, because many of the diseases discussed in this chapter can cause a severe sepsis-like syndrome in the neonatal period. Recent improvements in expanded newborn screening programs have made it possible to diagnose many inborn errors of metabolism within the first week of life, thereby reducing the uncertainty when acute metabolic dysfunction occurs or preventing it altogether.2 Accordingly, it is vital that the treating physician retrieve the results of the sick child’s newborn screen when the underlying diagnosis is not known.
In this chapter, we focus on straightforward treatment paradigms aimed at recognizing and reversing metabolic emergencies caused by the disorders listed in Table 114-1. Further discussion of the presentations of inborn errors of metabolism at various ages, and with different symptom complexes, as well as initial management is found in Chapter 134. Specific disorders are discussed in detail in the remainder of Section 11. In general, the goals of managing children with acute metabolic dysfunction are to
1. Stabilize cardiorespiratory function.
2. Determine the category of metabolic dysfunction (fatty acid oxidation, urea cycle, etc).
3. Eliminate the offending agent.
4. Promote anabolic metabolism.
5. Remove toxins (eg ammonia, organic acids).
6. Arrange specialized diagnostic testing.
One further consideration is that many children with inborn errors of metabolism do not survive their first episode of metabolic decompensation. Because these are genetic diseases, accurate counseling about recurrence risk is of the utmost importance for the family and depends on establishing a definitive diagnosis in the affected child. This may require procuring fresh tissues for enzymatic analysis after death.3 In critically ill children who do not respond to initial efforts to reverse metabolic dysfunction, it is recommended that a pathologist be consulted to plan for the possibility of rapid tissue harvest immediately after death.
CATEGORIES OF INBORN ERRORS OF METABOLISM AND MANAGEMENT OF ACUTE DECOMPENSATION
 DISORDERS OF FATTY ACID OXIDATION, KETOGENESIS, AND KETONE BODY UTILIZATION
DISORDERS OF FATTY ACID OXIDATION, KETOGENESIS, AND KETONE BODY UTILIZATION
A large number of diseases (discussed in detail in Chapter 150) result in impaired fatty acid oxidation.4 All of these disorders are inherited as autosomal recessive traits. Except for the most severe cases, children are well at baseline but cannot tolerate periods of fasting or increased metabolic demand (eg, fever, infection) when fatty acid oxidation would normally be required. The impairment of fatty acid oxidation results in hypoketotic hypoglycemia, acute liver dysfunction, rhabdomyolysis, and cardiac failure. Hypoglycemia paired with inadequate ketogenesis impairs bioenergetics in the central nervous system, leading to lethargy that can progress rapidly to coma. Since lipolysis and fatty acid transport are not impaired, fatty acids accumulate in hepatocytes, where they may exert toxic effects in addition to impairing energy generation. Hyperammonemia and lactic acidosis may occur due to secondary effects on liver metabolism. The most common of the fatty acid oxidation disorders, medium-chain acyl-CoA dehydrogenase (MCAD, see Chapter 150) deficiency, has an incidence of approximately 1:10,000 and presents with severe hypoglycemic episodes characterized by nausea, lethargy, and potentially sudden death.4,5
There are also several diseases that impair production of ketone bodies from the acetyl-CoA produced during fatty acid oxidation (see Chapter 151) or that impair the utilization of ketone bodies by extrahepatic tissues (the ketone utilization disorders; see Chapter 152). Clinically, the disorders of ketogenesis are similar to fatty acid oxidation defects, with acute episodes of fasting-induced hypoglycemia and inappropriately low ketones in the blood and urine. The ketone utilization defects present with episodes of vomiting and profound ketoacidosis, typically with normoglycemia.
The most important goal in acute management of these diseases is to reverse the catabolic state as quickly as possible. This is done most effectively by stimulating insulin release with a high glucose infusion rate (6–8 mg/kg per minute). Insulin and dextrose suppress lipolysis in the adipose tissue and suppress fatty acid oxidation in the liver and elsewhere. These actions curtail the supply of free fatty acids to the liver and reduce the accumulation of partially oxidized intermediates in the β-oxidation pathway. A 10% dextrose-based solution at 1.5 to 2 times the estimated maintenance rate should be used initially. If central access can be obtained, a higher dextrose concentration can be used to limit the volume of the infusion. Insulin infusions are helpful to augment endogenous insulin secretion in severely ill children. These interventions also benefit acutely ill children who are normoglycemic, because the increase in circulating free fatty acids may produce symptoms of hepatotoxicity prior to the decrease in blood glucose. For patients with these disorders requiring parenteral nutrition, intralipid mixtures must be avoided.
Table 114-1. Major Categories of Inborn Errors of Metabolism Causing Acute Metabolic Dysfunction
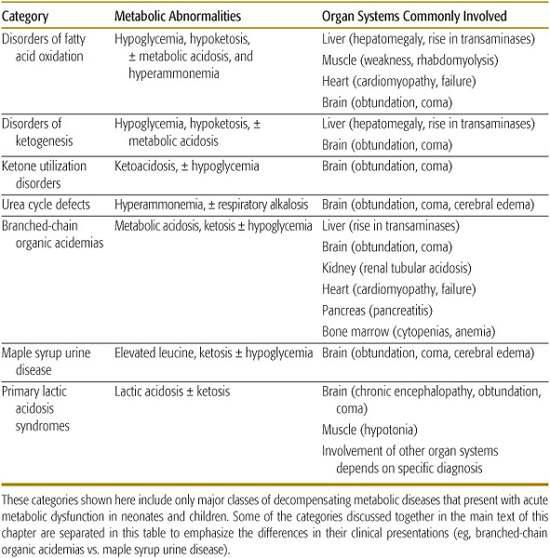
Table 114-2. Questions That Should Prompt Suspicion of an Inborn Error of Metabolism in a Critically Ill Child



