Acute Lymphoblastic Leukemia
James B. Nachman
Acute lymphoblastic leukemia (ALL) is the most common childhood malignancy, accounting for almost 25% of cancer diagnoses seen in children younger than age 15 years. Patients with more than 25% lymphoid blasts in the bone marrow are considered to have leukemia regardless of the presence of extramedullary disease. Patients who have an extramedullary lymphoid mass and 5% to 25% lymphoid blasts in the marrow are considered to have lymphoblastic lymphoma with marrow involvement. There are approximately 2400 new cases of ALL diagnosed in children and adolescents less than 20 years of age each year in the United States.1 The peak incidence of ALL is in the 2- to 6-year-old age group. The incidence of ALL appears to be highest in Hispanic children and lowest in African American children.2
Progress made in treating ALL has been remarkable. As recently as 50 years ago, cure rates for patients with ALL were less than 10%. In 2008, we expected to cure approximately 80% to 85% of patients with ALL.3-6 Patients are generally considered cured if they remain in remission more than 5 years from discontinuation of therapy. Chemotherapeutic principles and supportive care guidelines derived from the experience in treating ALL have had a major impact on the treatment of other types of cancer.
 BIOLOGY
BIOLOGY
In the developmental pathway of lymphocytes (see Fig. 449-1), maturation to the mature B-cell and T-cell state occurs without the necessity of antigenic stimulation—so-called antigen independent differentiation. When a B or T cell combines with its specific antigen, proliferation and differentiation occur, resulting in long-lived memory cells for antigen recognition and plasma cells which secrete antibody or T cells with various effector functions, such as cytotoxicity, B-cell help, cytokine secretion, and so on. In B-lineage acute lymphoblastic leukemia (ALL), approximately 98% of patients show a B-precursor immunophenotype. The rare patient with mature B-cell leukemia has a form of Burkitt lymphoma and requires different treatment. The various stages of B-cell development cannot be judged by cell morphology. Surface antigen expression is used to determine the stage of B-cell development. The earliest stage in B-cell development is the pro-B cell, which is characterized by surface expression of histocompatibility complex (HLA-DR) and the lack of expression of CD10 (common ALL antigen). The common precursor B cell expresses HLA-DR and CD10 but does not express cytoplasmic immunoglobulin. The pre-B cell expresses cytoplasmic immunoglobulin, and may or may not express CD10. The incidence of the various subtypes of B-precursor ALL is approximately 75% common B, 20% pre-B, and 5% pro-B.4 The pro-B immunophenotype is most often seen in infants. In T-ALL, cases are evenly distributed between the early, mid-, and late thymocyte state determined by the presence or absence of various T-cell antigens such as CD 2, 4, 5, 7, 8, and cytoplasmic CD3. Rarely, ALL cases may show a biphenotypic, bilineage, or natural killer cell immunophenotype.
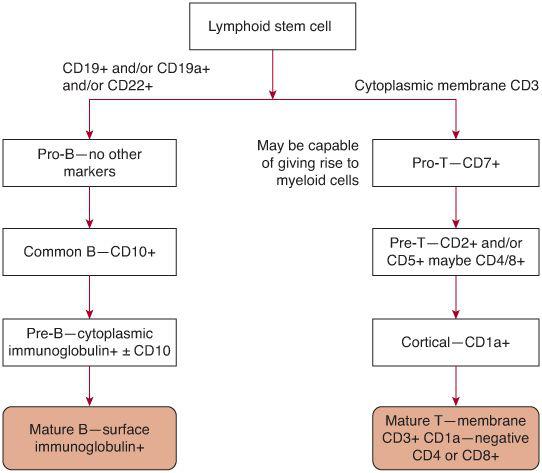
FIGURE 449-1. Development states of B- and T-cell leukemia in childhood.
To determine cytogenetic abnormalities in childhood ALL, metaphase preparations of leukemic cells are prepared and abnormalities in chromosome number and structure are determined. It is now well known that certain genetic abnormalities are not detected by routine chromosome analysis. Therefore, florescent in situ hybridization (FISH) is also performed on leukemic cell spreads. Most cases of B-lineage ALL show chromosomal abnormalities detected by either standard cytogenetic or molecular techniques. These abnormalities may include gains or losses of whole chromosomes, structural abnormalities involving individual chromosomes, and translocations involving 2 or more chromosomes. The two most common genetic aberrations in childhood B-lineage ALL are hyperdiploidy (usually > 51 chromosomes) and a translocation between chromosomes 12 and 21, often referred to as TEL – AML1 because of the genes involved in the breakpoints on chromosomes 12 and 21. Both of these genetic abnormalities are most common in children 1 to 9 years of age, show a common B immunophenotype, and are associated with a good prognosis.7 Hypodiploidy (< 44 chromosomes) and a translocation between chromosomes 9 and 22, the so-called Philadelphia chromosome, are rare genetic abnormalities associated with a poor prognosis.8,9 The incidence of Philadelphia-chromosome-positive ALL increases with age and may account for 5% of cases in patients 16 to 21 years of age. Other genetic abnormalities are not associated with either favorable or unfavorable prognosis. Approximately 25% of cases of pre-B ALL show a translocation between chromosomes 1 and 19.10 In T-cell ALL, it is uncommon to find cytogenetic abnormalities based on examination of chromosome spreads. However, molecular analysis often reveals recurring abnormalities. For example, mutations in the Notch gene occurs in approximately 50% of cases of childhood T ALL and appear to be associated with a good prognosis.11
 CLINICAL PRESENTATION
CLINICAL PRESENTATION
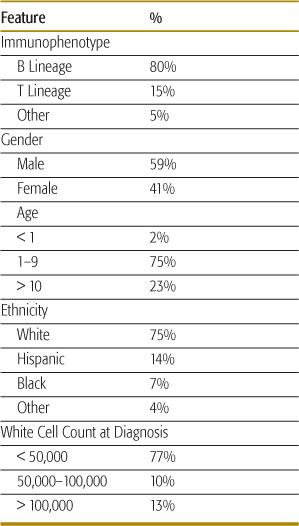
The demographic features of childhood acute lymphoblastic leukemia (ALL) are shown in Table 449-1.
Table 449-1. Demographics of Children and Adolescents with ALL
In many cases of childhood ALL, production of the various types of blood cells other than lymphocytes is seriously impaired. A low red cell blood count (RBC) count leads to pallor; decreased platelets lead to bruising, and a low neutrophil count may lead to fever as a sign of infection. The triad of fever, pallor, and bruising is a common presentation of ALL, particularly in younger children. In some younger patients, severe bone pain in the back and lower extremities with mild or no abnormalities in the blood count may be presenting symptoms of ALL. A number of patients with ALL present with lymphadenopathy and hepatospenomegaly, due to infiltration by malignant lymphocytes. Rarely, patients with ALL may present with respiratory compromise due to the presence of a large mediastinal mass (particularly more common in T-cell ALL). This is a medical emergency and requires immediate intervention to prevent respiratory arrest. Another rare presentation of ALL is that of a lytic bone lesion. Prior to performing a biopsy of any lytic bone lesion, a complete blood count should be obtained and a careful review of the peripheral smear should be carried out.
 DIAGNOSIS
DIAGNOSIS
Most patients with acute lymphoblastic leukemia (ALL) will show at least some blast cells on a peripheral blood smear. ALL blasts are characterized by a high nuclear cytoplasmic ratio, clumpy nuclear chromatin, and few small nucleoli. Even if blast cells are present in the peripheral blood, performing a bone marrow aspiration is highly recommended to obtain specimens for determination of immunophenotype by flow cytometric techniques and cyto-genetic analysis. Both immunophenotype and cytogenetics are necessary for treatment stratification.7 Most cases of ALL are either of clear Bor T-lineage origin. Either B- or T-lineage ALL cases may show aberrant expression of myeloid antigens.12 If myeloid antigen(s) are present, a scoring system can be used to determine whether or not the leukemia has biphenotypic features, which is generally associated with a poor prognosis. This does not appear to be the case in acute myeloid leukemia, which expresses lymphoid antigens. Approximately 80% of cases of childhood ALL are B lineage, whereas 15% are T lineage. T-lineage cases account for approximately 10% of cases in children ages 1 to 9 years and approximately 20% of cases in adolescents and young adults. In any patient with ALL, a lumbar puncture should be performed to assess whether blasts are present in the cerebrospinal fluid. The initial puncture should be done by an experienced physician and younger patients should be under sedation to minimize the likelihood of a traumatic tap. More than 10 RBC/μL in cerebral spinal fluid (CSF) is an adverse prognostic factor. In a non-traumatic tap, the presence of 5 or more white blood cells and the presence of blasts in a cytocentrifuged specimen are needed to make a diagnosis of central nervous system involvement (CNS-3). Patients with less than 5 white blood cells in the cerebrospinal fluid but with leukemic blasts on the cytocentrifuge specimen are given the designation CNS-2. Patients with symptoms referable to the CNS should always have either a head computerized tomography (CT) scan or magnetic resonance imagine (MRI) prior to having a lumbar puncture.
 PROGNOSTIC FACTORS
PROGNOSTIC FACTORS
A large number of prognostic factors have been identified that are utilized to determine treatment. Prognostic factors can generally be divided into three groups: disease-related factors, host-related factors, and treatment response factors.
Age is an important prognostic factor in acute lymphoblastic leukemia (ALL).13 Generally, patients age 1 to 9 years have a better prognosis than those under 1 year of age or those older than 10 years. This is in part related to the significantly higher incidence of favorable cytogenetic subtypes of ALL (hyperdiploidy and the 12/21 translocation) in the 1-to-9-year age group. Between 50% and 60% of children in the 1-to-9-year age range will have either hyperdiploidy or translocation.12,21 Infants less than 1 year of age have a high incidence of abnormalities associated with the 11q23 chromosome locus.14 The most common abnormality is translocation.4,11 However, even if patients are matched for cytogenetic abnormalities, patients 1 to 9 years fare better than older patients.
White blood cell (WBC) count is another important prognostic factor in ALL. It is more important in B-lineage as opposed to T-lineage ALL. Many studies utilize an arbitrary cutoff of 50,000 WBC/μL at diagnosis in determining risk group assignment. Extreme hyperleukocytosis (> 200,000) is often associated with unfavorable cytogenetics and generally carries a poor prognosis.
The combination of age and white blood cell (WBC) count was one of the initial prognostic factors used to determine outcome. Patients 1 to 9 years of age and WBC less than 50,000/μL had a better prognosis than patients 1 to 9 years and more than 50,000 WBC or than patients more than 10 years of age at the time of diagnosis.
Race has been evaluated as a prognostic factor in many studies, but with modern treatment, race tends not to be an important prognostic factor.15 However, it is clear that racial background may play a role in determining clearance and toxicity associated with various chemotherapeutic agents.
The presence of central nervous system leukemia at the time of diagnosis (> 5 WBC/μL and the presence of blast cells in the cerebrospinal fluid, or presence of cranial nerve palsies) is a poor prognostic factor. Initial testicular involvement in males is also thought to be a high risk factor.16 Hepatosplenomegaly and other extramedullary involvement at diagnosis has no independent prognostic significance.
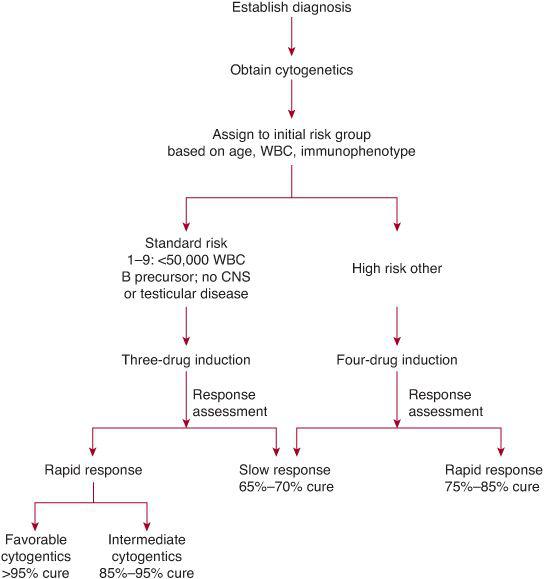
Pretreatment factors were initially used to determine risk group and treatment assignment. However, rapidity of response to induction chemotherapy is currently recognized as the most important factor determining outcome.17 Within any subgroup defined by age, WBC, and/or cytogenetics, patients with no or minimal residual disease (MRD) at the end of induction have a significantly better outcome compared to patients with higher levels of MRD. Therefore, children with ALL are assigned to an induction treatment based on age, white blood cell count, and immunophenotype and then treatment is modified at the end of induction based on rapidity of response and cytogenetics (See Fig. 449-2 and Table 449-2). Initially, morphologic response in peripheral blood following a 7-day treatment prophase with prednisone alone was shown to have prognostic significance.17 Patients with less than 1000 blasts/μL in peripheral blood on day 8 had a cure rate greater than 70%, whereas patients with greater than 1000 blasts/μL had a cure rate of approximately 30%. Patients with more than 25% blasts in the bone marrow on day 8 of induction therapy had a significantly worse prognosis compared to patients showing less than 25% blasts on the day-8 marrow examination. Patients showing slow early response were assigned to more aggressive treatment strategies and in some studies this led to an improvement in outcome.18
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


