- Brain development
- Malformations of the central nervous system
- Disorders of head size and shape
- Intracranial haemorrhage
- Periventricular leukomalacia
- Neonatal stroke
- Hypoxic–ischaemic encephalopathy
- Neonatal convulsions
- Neonatal hypotonia (‘floppy infant’)
Introduction
Development of the brain is not only a complex but a continuous process starting from very early fetal life, right through gestation and continuing through the first few years of life. The milestones of central nervous system (CNS) development include formation of neural tube and hemispheres, neuronal proliferation, migration and wiring of the brain, synaptogenesis and mylenation. The developing brain is also an extremely vulnerable organ and is subject to a wide range of insults, both in nature and timing, that may alter its structure and function. Advances in diagnostic techniques such as neuroimaging and molecular biology have improved our understanding of the mechanism of perinatal brain injury and this in turn should help to predict long-term neurodevelopmental outcomes.
Brain Development
Development of the brain is a continuous process starting from very early fetal life, right through gestation and until the end of the first decade of life. The pattern of brain growth and development is illustrated in Fig. 22.1.
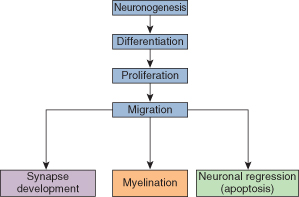
Neuronogenesis
The first neural tissue appears at about 18 days with the neural crest, from which the neural tube develops. There is a phase of rapid neuronogenesis occurring from 4 to about 18 weeks of gestation.
Differentiation
The primitive neural cells differentiate into the different populations of cells, both neurons and glia, that make up the mature brain. This process occurs mainly from weeks 4 to 18.
Proliferation
There is a vast increase in the number of neuronal cells up to about 18 weeks of gestational life, most of which die as part of the neuronal regression process (see below).
Neuronal Migration
Neurons are produced deep in the brain and migrate to the cortex and other sites. Final migration of neurons has been achieved by 25 weeks of gestation.
Neuronal Regression
The process of apoptosis ensures that only neurons that have achieved a functional capacity within the nervous system survive. Most neurons, during the course of migration, do not acquire this function and regress as part of normal brain development.
Synapse Development
Full function in the brain depends on the dendrites of each neuron developing and making many connections (on average each neuron is in contact with 10,000 other neurons). These connections are called synapses. Establishing new synaptic contacts is part of the process of development and learning.
Myelination
Glial cells are associated with the process of myelination and there is rapid growth of these cells from 24 weeks as they migrate to their final sites. Myelination is not complete until the age of about 12 years.
Factors adversely influencing brain growth and development may operate at different times. Community-based studies of disabled children, relating the timing of the brain insult to either prenatal, perinatal or postnatal events, show that prenatal insults account for more disability than perinatal and postnatal causes together. The approximate proportions for neurological handicap in a community are shown in Table 22.1. Table 22.2 lists the relatively common insults that may cause prenatal, perinatal or postnatally acquired disability.
Table 22.1 Estimated timing of events that cause neurological handicap in a community
| Prenatal | 70–75% |
| Perinatal | 20% |
| Premature | 60% |
| Full-term | 40% |
| Postnatal | 5–10% |
Table 22.2 Causes of neurological disability related to the timing of onset of the insult
| Prenatal | Perinatal | Postnatal |
| Down’s syndrome | Birth asphyxia | Hypothyroidism |
| Neural tube defects | Intracranial haemorrhage | Meningitis |
| Other chromosomal disorders | Periventricular leukomalacia | Inborn errors of metabolism |
| Viral infection | Ototoxic drugs | Trauma |
| Intrauterine growth restriction | Kernicterus | |
| Toxins and drugs | Hypoglycaemia |
The brain is not a homogeneous organ. Specific areas of the brain grow at different times and rates. Thus, growth restriction at any one time (e.g. due to malnutrition) distorts the general growth of the brain and may selectively affect a particular area. Unlike general body growth, the brain has only one opportunity to develop properly and thus interference with growth at a particular time in development may be irreversible.
Malformations of the Central Nervous System
Abnormalities of the brain can be classified as malformations (a developmental defect in which the brain was never normal) and deformations, where an external insult has affected normal brain development causing an abnormality in subsequent structure.
Malformations of the CNS apparent at birth result from abnormalities in CNS development. These can be divided into two groups: disorders of dorsal induction and disorders of ventral induction.
- Dorsal induction refers to the formation and migration of the neural tube, with subsequent development of the anterior tube into the primitive brain structures. These processes occur during the third and fourth weeks of gestation. Disorders occurring at this time include anencephaly, encephalocoele, myelomeningocoele and meningocoele. These are collectively known as neural tube defects (NTDs), and the incidence of babies born with these disorders has fallen markedly over the last 15 years.
- Ventral induction refers to development at the ventral end of the neural tube, and particularly cleavage into bilateral hemispheres and ventricles, with thalamic and hypothalamic growth. These processes occur mainly in the fifth and sixth weeks of gestation. The commonest disorder occurring at this time is holoprosencephaly, which may be associated with abnormalities in facial development.
Neural Tube Disorders (NTD)
NTDs in general, and spina bifida in particular, have become much less common in recent years. There are two main reasons for this: periconceptual folate supplementation and antenatal fetal screening. In some women the risk of a baby with NTDs is increased. This includes families with a history of an affected child, or where the mother herself has the condition. A number of anticonvulsant drugs administered to the mother (particularly sodium valproate) are associated with a considerably increased risk.
It is now known that folic acid is an important substrate for normal early neural tube development, and periconceptual supplementation of at-risk women with folic acid reduces the incidence of NTDs by approximately 75%. As it is not possible to know which women are at increased risk until their first baby is born with spina bifida, it is now recommended that all women intending to become pregnant take regular folate for 3 months before conception.
The second factor in the falling incidence of NTDs is early fetal ultrasound to detect congenital spine or brain abnormalities. In many developed countries virtually all pregnant women are screened at about 18 weeks of gestation. The detection of a seriously abnormal fetus offers the opportunity for the parents to consider terminating the pregnancy.
Anencephaly
In this condition the forebrain is absent; it occurs as the result of an insult before 24 days’ gestation. With routine fetal scanning and termination of pregnancy this condition is now rarely seen at birth. Anencephaly is incompatible with life and results in stillbirth or neonatal death.
Encephalocoele
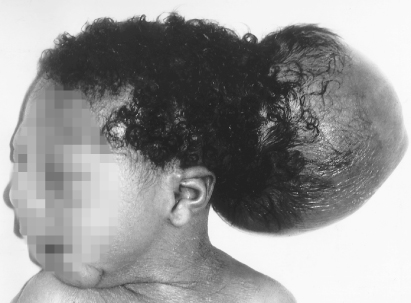
In this condition there has been failure of midline closure of the skull, usually with herniation of the brain. Up to 80% of cases occur in the occipital region (Fig. 22.2). This lesion occurs about 28 days after conception. The prognosis depends on the amount of brain in the sac. If the infant is microcephalic with a large encephalocoele, the prognosis is very poor. Neurosurgery is necessary to close the defect.
Figure 22.2 Occipital encephalocoele. This infant was operated on. Although he has severe visual impairment, at 2 years he was otherwise normal.
Spina Bifida
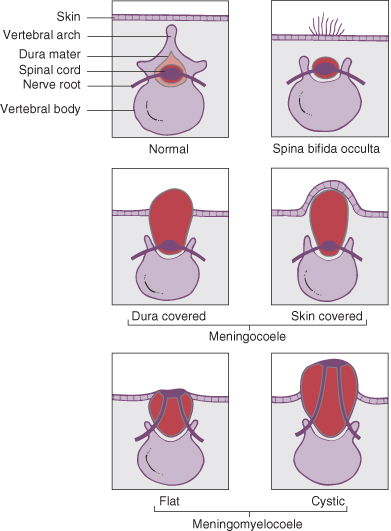
This is a developmental failure of fusion of the vertebral column, often with an external protrusion of the meninges and cord. A meningeal sac of cerebrospinal fluid (CSF) with normal underlying spinal cord is referred to as a meningocoele, and if there is associated abnormality of the cord it is a myelomeningocoele. The abnormalities produced may be classified according to their severity.
Spina Bifida Occulta
In this condition the vertebrae are bifid but there is no meningocoele or myelomeningocoele sac (Fig. 22.3). This abnormality is seen in 10% of the population and is usually of no clinical significance. In a small proportion the spinal cord may be tethered and with growth becomes stretched, causing irreversible neurological signs in the lower limbs and bladder. This condition should be suspected if there are lesions over the midline of the lower back. Such lesions include:
- a deep sinus
- a naevus
- a tuft of hair
- a soft fatty swelling referred to as a lipomyelomeningocoele – this is particularly likely to be associated with later neurological signs.
All newborn infants with any of these clinical features should be investigated with real-time ultrasound and, if spinal cord tethering is suspected, referred to a neurosurgeon. The prognosis is much better with early repair before the onset of neurological symptoms.
Spina Bifida Cystica
This includes meningocoeles and meningomyelocoele (Fig. 22.3). The incidence of this lesion in liveborn infants is now about 1/1000 births, but is considerably higher in some parts of the world.
Meningocoele
These account for 20% of spina bifida cystica lesions. In this condition there is no herniation of nervous tissue, and consequently no neurological deficit. There is a risk of meningitis if the sac leaks CSF.
Myelomeningocoele
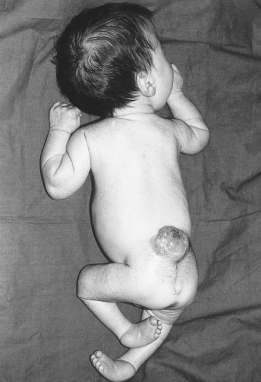
These account for 80% of spina bifida cystica lesions. They are associated with herniation of nervous tissue and permanent neurological deficit (Fig. 22.4). The clinical feature are listed in Box 22.1
- Site of lesion. About 70% are lumbosacral
- Covering of sac. Usually meninges, but occasionally the sac is ruptured, leading to CSF leakage and consequent risk of meningitis
- Neurological examination:
- Hydrocephalus. Ninety percent of infants with meningomyelocoeles have an associated abnormality of the brain and skull base, referred to as the Arnold–Chiari type II malformation. This includes prolapse of the medulla, cerebellum and fourth ventricle through an abnormal foramen magnum into the cervical canal. Not all infants develop hydrocephalus, but most have ventricular dilatation on ultrasound scans. Progressive enlargement of the head may occur with advancing age
- Orthopaedic abnormalities. These are common and include talipes (see Figure 22.4), dislocated hips, kyphosis, scoliosis and contractures of the lower limbs
- Miscellaneous abnormalities. These include renal, cardiac and visceral defects and chromosome disorders
Investigations
The neural arches of the vertebrae are poorly mineralized at birth, and spinal radiography is of little value except for the assessment of scoliosis. Ultrasound examination of the spine in the newborn period allows visualization of the spinal cord: if it does not move with respiration, this suggests that there may be tethering.
Management
A careful assessment of the newborn infant by appropriate specialists is necessary before a definite treatment plan can be formulated. Treatment is always discussed with the parents, whose wishes should be considered. Many centres use Lorber’s criteria for conservative treatment. Lorber followed a large number of babies with meningomyelocoele, and identified the following bad prognostic criteria:
- total paralysis of the legs
- thoracolumbar or thoracolumbosacral lesions
- severe kyphoscoliosis
- hydrocephalus at birth
- other major congenital malformations
If one or more of these features are present at birth, he recommended conservative management. This consists of nursing care only, but does not rule out subsequent reappraisal of the need for neurosurgery. Some centres close the skin lesion routinely even if opting for palliative care.
If active treatment is indicated, the following approach would be adopted:
- Early neurosurgery. Closure of the sac within 24 h of birth, with ongoing assessment for hydrocephalus and insertion of a ventriculoperitoneal shunt, if indicated.
- Orthopaedic assessment and treatment as necessary.
- General surgical treatment. Urinary incontinence is a major problem, and girls usually require an ileal conduit, whereas boys may be managed at least initially with a penile collecting system or intermittent catheterization. The aim of treating faecal incontinence is to produce a firm stool to prevent soiling and faecal impaction.
- Supportive care. Pressure sores need to be prevented by careful positioning and skin care. Psychological and social problems are common and parents need careful support and counselling.
- Genetic counselling. This will be necessary for future pregnancies.
Screening for Neural Tube Defects
There are two main aspects of screening for NTDs:
- Alpha-fetoprotein (AFP). Blood is taken from the mother at 14–18 weeks’ gestation and the level of AFP is measured. AFP in the first trimester normally increases with gestational age, and an accurate assessment of the duration of pregnancy is essential in assessing the significance of the AFP level. Those women with high serum levels should have repeat samples taken 1 week later. Only 10% of pregnancies complicated by a high serum AFP level are associated with NTDs. Multiple pregnancies, exomphalos and other abnormalities may cause high levels. AFP is raised in 90% of cases of anencephaly, and in most cases of open myelomeningocoele. A skin-covered lesion will not have raised levels.
- Ultrasound screening. Various abnormalities are assessed, including careful examination of the lower spine for a skin defect and examination of the skull base for the ‘banana’ sign (a feature of the Arnold–Chiari malformation) or a ‘lemon’ sign involving the frontal bones.
Disorders of Ventral Induction
Holoprosencephaly
This is a rare condition where there is a failure of midline fusion of the CNS. There may also be midline defects of the eyes, lips and palate. Half of these lesions are associated with abnormal chromosomal patterns, most often trisomy 13 or 18. The prognosis for normal development is hopeless.
Holoprosencephaly may present as complete alobar forms or as partial forms, referred to as lobar or semilobar, and the diagnosis is made on ultrasound or CT/MRI examination. The appearances of agenesis of the corpus callosum and septo-optic dysplasia may be confused with holoprosencephaly if care is not taken. If septo-optic dysplasia is suspected, assessment of the baby’s vision and hypothalamic/pituitary function should be made.
Disorders of Head Size and Shape
Microcephaly
This is defined as an occipitofrontal head circumference more than two standard deviations below the mean for the infant’s gestational age. The child may show microcephaly in proportion to weight and length (symmetrical growth restriction) or, more ominously, have a small head but a normally grown body. Microcephaly may be primary or secondary. Primary microcephaly results from disorders of neuronal proliferation (lissencephaly) or cellular migration (such as in Zellweger’s syndrome). Secondary microcephaly implies normal growth up to a point when a major insult has occurred, after which brain growth fails. Table 22.3 lists various causes of microcephaly. The prognosis depends on the underlying cause and is generally poor except for familial cases.
Table 22.3 Causes of primary and secondary microcephaly
| Primary | Secondary |
| Familial | Intrauterine growth retardation |
| Autosomal recessive | Meningitis |
| X-linked recessive | Hypoglycaemia |
| Chromosomal | Asphyxia |
| Trisomy 13 | Periventricular leukomalacia |
| Trisomy 18 | |
| TORCH infections | |
| Maternal phenylketonuria | |
| Lissencephaly | |
| Fetal alcohol syndrome |
The underlying cause should be diagnosed and treated wherever possible. The prognosis is usually poor and treatment is supportive.
Craniostenosis (Craniosynostosis)
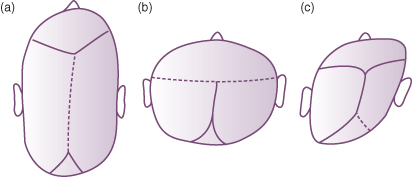
In this condition the skull sutures fuse prematurely resulting in distortion of head shape (Fig. 22.5). The most common form of craniostenosis is premature closure of the sagittal suture, giving a scaphocephalic head shape (boat shaped, see Chapter 6). Premature closure of the coronal suture leads to a turricephalic head (towering head), and closure of one lambdoid suture results in plagiocephaly (oblique head). Positional plagiocephaly also results from the baby lying on one side of the head with resultant distortion in shape. Autosomal dominantly inherited craniofacial deformities which are associated with some other abnormalities include:
- Apert’s syndrome (acrocephalosyndactyly)
- Crouzon’s syndrome (craniofacial dysostosis)
- Carpenter’s syndrome (acrocephalopolysyndactyly).
Figure 22.5 Premature suture closure leading to craniostenosis. (a) Scaphocephaly (sagittal suture); (b) turricephaly (coronal suture); and (c) plagiocephaly (single lambdoid suture).
Management
Skull radiographs may confirm the clinical suspicion but CT scan provides better assessment of the state of the cranial sutures. Surgery is indicated if premature fusion of the sutures causes raised intracranial pressure. Surgery for purely cosmetic reasons is not indicated except in very select cases.
Macrocephaly (Large Head)
Macrocephaly is the term used to describe a large head where head circumference is more than two standard deviations above the mean. It can result from a variety of causes which can be broadly subdivided into
- enlargement of the skull bones
- enlargement of brain itself (megacephaly)
- enlargement of CSF spaces (hydrocephalus)
- enlargement of other structures (such as brain neoplasm or intracranial haematoma).
A large head can be an isolated finding and familial or associated with disorders of growth (Soto’s syndrome). There are a number of genetic conditions which are also known to be associated with macrocephaly, for example Beckwith–Wiedemann syndrome, neurofibromatosis type 1 and fragile X syndrome.
Hydrocephalus
Hydrocephalus is caused by an imbalance between the production and the absorption of CSF, with resultant dilatation of the cerebral ventricles. This can also result from obstruction to CSF circulation. The term macrocephaly or megalencephaly should not be confused with term hydrocephalus.
Classification
Non-Obstructive Hydrocephalus
In this type there is no interference with CSF flow. The excessive production of CSF is usually due to a papilloma of the choroid plexus.
Obstructive Hydrocephalus
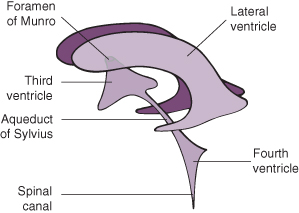
See Fig. 22.6 for site of obstruction. Obstructive hydrocephalus can be divided into non-communicating and communicating.
Figure 22.6 Diagram to show intracerebral drainage of cerebrospinal fluid.
Reproduced from Levene 1987, with permission of Churchill Livingstone.
Non-Communicating
There is little or no communication between the ventricles and the subarachnoid space. There are three common sites of obstruction:
- Aqueduct of Sylvius (due to stenosis or atresia).
- Occlusion of the foramina of Luschka and Magendie as a result of basal adhesions. This is the commonest site of blockage and is usually due to intraventricular haemorrhage.
- Arnold–Chiari type II malformation secondary to spina bifida cystica.
Communicating
CSF can escape from the intracranial system via the foramina of Luschka and Magendie, but cannot be absorbed at the arachnoid granulations situated over the surface of the brain. This is usually due to arachnoiditis following either meningitis or intraventricular haemorrhage (IVH).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


