- Development of the gastrointestinal tract
- Malformations
- Abdominal wall defects
- Congenital ascites
- Necrotizing enterocolitis
- Short bowel syndrome
- Rectal bleeding
Introduction
Normal structural and functional development of the gastrointestinal tract is essential for life. Malformations can occur anywhere from the mouth to the anus. Many of these can now be identified on antenatal ultrasound and there are some specific abnormalities that only present in the neonatal period. Normal coordination of suck and swallow does not appear until 34 weeks’ gestation and as a result feeding difficulties are one of the most common clinical problems encountered in the neonatal nursery. This chapter aims to give a broad overview of the specific conditions that affect the gastrointestinal tract in the neonatal period.
Development of the Gastrointestinal Tract
The gastrointestinal (GI) tract develops 4 weeks after conception as a tube from mouth to cloaca. The lip is normally fused by 5 weeks and the palate by 8–9 weeks. Part of the foregut differentiates into trachea and oesophagus, and disorders of development at this stage cause oesophageal atresia, usually with tracheo-oesophageal fistula. The lower bowel initially opens into the yolk sac and later forms the vitello-intestinal duct. The midgut forms a loop, which protrudes from the abdominal cavity and then re-enters the abdomen after turning through 270°. Failure of the bowel to re-enter the abdomen causes exomphalos, and failure to twist leads to malrotation. During the sixth week of gestation a septum separates the cloaca into rectum and urogenital sinus. The gut then ruptures through the perineum to form an anus. The liver and pancreas develop from the gut endoderm at the same time as the duodenum is formed. The gut is fully differentiated by 20 weeks.
Functional Development
Motility
Bowel motility is present from 16 weeks’ gestation but is not fully functional until 36 weeks. The passage of meconium in utero is rare before 34 weeks’ gestation. Functional intestinal obstruction or paralytic ileus are common findings in premature infants due to this disorganized bowel motility.
Swallowing
The fetus begins to swallow liquor at around 16 weeks’ gestation. This is an important factor in the regulation of amniotic fluid volume (see Chapter 1). If the upper gastrointestinal tract is not patent then polyhydramnios will develop. Although the preterm infant can swallow, nutritive suck and swallow does not start to become coordinated until 34 weeks’ gestation. Almost all preterm infants will require nasogastric or orogastric tube feeding until their coordinated suck–swallow is established. Gastro-oesophageal reflux is common, especially in the preterm infant, because of low gastro-oesophageal sphincter pressures and transient relaxations of the sphincter.
Carbohydrate Absorption
Disaccharidase activity in the small bowel is low at term and gradually increases to mature levels by 10 months of age. In the preterm baby maltase is the first disaccharidase to reach reasonable activity, followed by sucrase and then lactase. Lactase deficiency is common before 30 weeks’ gestation, with consequent lactose intolerance in these infants.
Fat Absorption
Bile salts are essential for fat absorption but are themselves not readily absorbed. Fat malabsorption in the newborn may occur because of reduced bile salts. In infants below 1300 g birthweight, 70–75% of dietary fat is absorbed. Premature infants are better able to absorb polyunsaturated fats (present in excess in human milk) than saturated fats. Some degree of physiological steatorrhoea is therefore normal in preterm infants. Premature infants cope better with the absorption of medium-chain triglyceride fats than long-chain fatty acids.
Secretions
Gastric acid secretion does not occur to any significant degree before 32 weeks’ gestation. Pancreatic secretions of lipase are adequate for dietary needs by full term, but trypsin is often deficient, resulting in relative protein malabsorption.
Malformations
Malformations of the gastrointestinal tract can occur anywhere from the mouth to the anus.
Cleft Lip and Palate
Cleft lip and palate are the most common congenital anomalies of the craniofacial region. Cleft lip or palate may occur in isolation but they are found together in up to 70% of cases. The lip and palate normally fuse by about 6 weeks’ gestation. Cleft lip may be unilateral (70% on left side) or bilateral. Clefts in the palate can have one of three forms:
- Complete, involving the hard and soft palate, and may involve the alveolar margin. It may be unilateral, bilateral or involve the midline.
- Submucosal, a bony defect completely covered by mucosa, and often a bifid uvula.
- Simple clefts of the soft palate only.
Cleft lip and palate have been associated with maternal anticonvulsant therapy, and also occur in the fetal alcohol syndrome. Many clefts will be diagnosed on antenatal ultrasound.
Management
Management is best undertaken by a multidisciplinary team involving plastic surgeons; ear, nose and throat surgeons; orthodontists; speech pathologists; psychologists; audiologists and specialist nurses. If the diagnosis is made antenatally, arrangements should be made to talk to someone from the team before delivery.
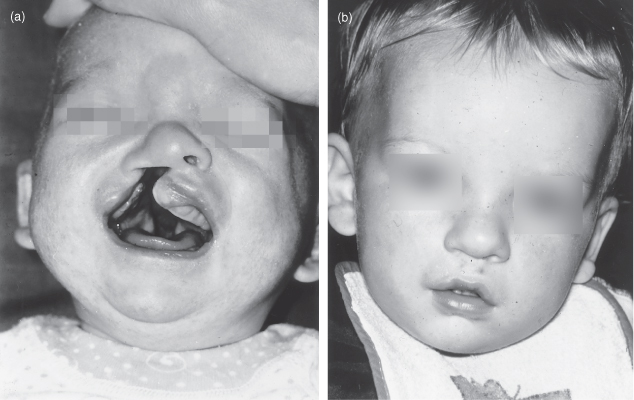
The infant often has an unattractive appearance at birth and time must be spent with the parents talking to them about the condition. Photographs of treated cases showing the preoperative and postoperative appearances are particularly helpful in allaying parental anxieties (Fig. 17.1).
Figure 17.1 Cleft lip. At birth the infant has a right-sided cleft lip (a). The same infant following repair (b).
Feeding is usually a problem, particularly if there is a cleft palate, because the baby is unable to achieve adequate suction, with resultant regurgitation of milk through the nose. Aspiration of milk can cause recurrent pneumonitis. Feeding can often be successfully facilitated with the help of a speech therapist. A special teat may be required. The careful use of a squeeze bottle aids suction in many infants. In some cases the fitting of an acrylic dental obturator around the edges of the palate is helpful.
Most surgeons prefer to repair the lip in the first 6–12 weeks of life, but very early closure in the first week is recommended by some. Surgical closure of the palate is normally attempted at 9–12 months before speech has developed. Long-term complications of cleft lip and palate are listed in Table 17.1.
Table 17.1 Long-term complications of cleft lip and palate
| Speech and language | Problems include nasal escape and articulation. A speech therapist should always be closely involved in the management of these children |
| Hearing | Eustachian tube function is usually impaired, predisposing the child to middle ear infections. Regular hearing assessment and ENT supervision are essential |
| Dental | Tooth development is often delayed and there may be malocclusion |
| Local ulceration | Ulceration may be due to poorly fitting acrylic plates |
Reoccurrence Risk
It is usually of polygenic inheritance and has a higher frequency in some families. Cleft palate is associated with DiGeorge syndrome which is autosomal dominant with a 50% reoccurrence risk. The risk of recurrence in subsequent pregnancies is about 3–5%. It is more common in the pregnancies of older mothers. Recurrence risk counselling is affected by whether the lesion is unilateral or bilateral, is associated with cleft palate, and if other first-degree relatives are affected. Cleft lip with or without cleft palate is associated with a broader pattern of altered morphogenesis in 35% of cases.
Intestinal Obstruction
The causes of obstruction may be classified depending on the site of blockage (large or small bowel) or whether it is anatomical or functional. Causes of intestinal obstruction are listed in Table 17.2. On occasions it is impossible to be sure of the diagnosis or even the level of obstruction, and laparotomy may be the only way of making a diagnosis in order to treat the condition effectively. Intestinal obstruction occurs in about 1/1000 babies. If the obstruction is in the upper gastrointestinal tract it is often associated with maternal polyhydramnios. If more than 25 mL of fluid is aspirated from the stomach after birth, intestinal obstruction should also be suspected.
Table 17.2 Types of intestinal obstruction in the newborn
| Congenital | Acquired | Functional |
| Intrinsic | ||
| Atresias – oesophageal, duodenal, small bowel, colonic | Necrotizing enterocolitis | Hirschsprung’s disease |
| Intussusception | ||
| Peritoneal adhesions | Meconium plug syndrome | |
| Stenoses | ||
| Anorectal malformation | Ileus | |
| Meconium ileus | Peritonitis | |
| Enteric duplications | ||
| Extrinsic | ||
| Volvulus | Intestinal pseudo-obstruction syndrome | |
| Peritoneal bands | ||
| Annular pancreas | ||
| Cysts and tumours | ||
| Incarcerated hernia | ||
Clinical Features
The infant with intestinal obstruction may present with some or all of the following features:
- Bile-stained vomiting. This is a very important sign that must not be ignored. A history of bile-stained vomiting demands immediate investigation as it suggests obstruction is below the second part of the duodenum. Sporadic bile-stained vomiting suggests partial obstruction caused by malrotation, duplication or annular pancreas.
- Abdominal distension. This may not be prominent with a high obstruction.
- Visible peristalsis.
- Delayed passage of meconium. In cases of low obstruction there may be no passage of meconium at all. With a high obstruction meconium may be passed for a day or two. A changing stool is never seen with congenital bowel obstruction. Anal atresia should be recognized early at routine examination (see below).
- Dehydration. The infant may present with dehydration and collapse as the result of excessive vomiting.
Investigations and Management
The following investigations and management options apply in cases of suspected intestinal obstruction.
- Obstetric ultrasound as early as 17–19 weeks might reveal suspicious features such as polyhydramnios, ‘double bubble’ or echogenic bowel.
- Initial abdominal radiographs should be plain films with infant in the supine and left lateral positions:
- Withhold feeds, insert IV and nasogastric replogle (double-lumen) tube.
- Consult paediatric surgeon if obstruction confirmed or suspected.
- If diagnosis is in doubt or meconium ileus suspected, a contrast enema is performed: microcolon suggests small bowel atresia or meconium ileus.
- An upper gastrointestinal contrast study is performed if plain film and contrast enema are non-diagnostic.
Malrotation (Volvulus Neonatorum)
This results from incomplete fixation and rotation of the bowel after it returns to the fetal abdominal cavity from the yolk sac between the eighth and tenth weeks of gestation. The three features of malrotation are:
- the duodenojejunal junction is at or to the right of the vertebral column
- the ileocaecal junction is near the midline and higher than normal
- the bowel is abnormally fixed by avascular (Ladd’s) bands, which cross the second part of the duodenum.
These abnormalities cause the small bowel mesentery, in which the superior mesenteric artery lies, to be abnormally mobile and to twist around its axis, leading to a volvulus with rapid impairment of gut blood flow. This may cause intestinal obstruction in one of two ways:
- strangulation obstruction: the superior mesenteric artery supplying blood to the bowel is occluded
- Ladd’s bands obstruct the second part of the duodenum.
Midgut volvulus can occur at any time but 80% of cases occur in the neonatal period or in utero.
Diagnosis
This lesion characteristically produces episodic obstruction with abdominal distension, bile-stained vomiting, pallor and a vague abdominal mass in an infant who was previously tolerating feeds well. The baby may rapidly proceed to shock.
Plain radiography of the abdomen in the erect position may show a characteristic double bubble, but gas is seen beyond the duodenum. Contrast studies may show a corkscrew duodenum or an abnormally situated subhepatic position of the caecum. Ultrasound scan may show an abnormal relationship between the superior mesenteric artery and vein strongly suggestive of malrotation.
Treatment
Laparotomy needs to be performed urgently to untwist and relieve the volvulus. It may be difficult to exclude volvulus clinically and, in view of the rapidity with which bowel infarction occurs, an early laparotomy is advisable in suspected cases.
Pyloric Stenosis
Vomiting (often projectile) is the predominant symptom in this condition which occurs between birth and 6 months of age. It most commonly presents between weeks 3–5 as vomiting after feeding. The vomitus contains partially digested milk but no bile. Gastric peristalsis may be seen. Preterm infants will often present with pyloric stenosis prior to hospital discharge. The condition is due to hypertrophy of the pylorus. A pyloric mass is sometimes palpable in the right upper quadrant, particularly in cases where diagnosis has been delayed. There is often a family history, and boys are affected four times more often than girls. The cause of the hypertrophy is not known, but may be related to stress in a genetically susceptible infant. Incidence is 1/1000 to 1/3000. The risk for subsequent siblings is about 7%. Diagnosis is made using ultrasound examination and finding the pylorus 3 mm or more in thickness.
Treatment is surgical following adequate restoration of electrolyte and fluid balance and correction of metabolic alkalosis. At surgery the muscle fibres of the hypertrophied pylorus are incised down to the mucosa. This is known as Ramstedt’s procedure and is performed via a periumbilical incision or laparoscopically.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


