Metabolic Abnormalities
William W. Hay Jr.
DISORDERS OF GLUCOSE METABOLISM
Normal postnatal glucose homeostasis is established by increased glucose production and glucose utilization.1 Factors that promote glucose production and release into the circulation include catecholamines and glucagon, which activate glycogenolysis. A high glucagon-to-insulin ratio, which induces synthesis and activity of the enzymes, is required for gluconeogenesis. Once normal feedings are established, glycerol and amino acids continue to fuel gluconeogenesis while dietary fatty acids activate the enzymes responsible for gluconeogenesis. Additionally, galactose derived from hydrolysis of milk sugar (lactose) in the gut increases hepatic glycogen production for sustained between-feeding hepatic glucose release from glycogen breakdown. Feedings also induce production of intestinal peptides, or incretins, that promote insulin secretion. Insulin decreases hepatic glucose production and increases glucose utilization for energy production and storage as glycogen. These opposing conditions of glucose production and utilization continue in response to normal feed-fast cycles, regulating normal plasma glucose concentrations.2
HYPOGLYCEMIA
Glucose is the major source of energy for organ function. All organs use glucose, and glucose deficiency leads to impaired cardiac performance, cerebral energy failure, hepatic glycogen depletion, and muscle weakness.3 Cerebral glucose metabolism accounts for as much as 90% of total glucose consumption in the newborn. Thus, maintenance of glucose delivery to all organs, particularly the brain, is an essential physiological function.4 Although alternate fuels can substitute for glucose metabolism, concentrations of these substances often are low in newborn infants, especially preterm infants. Newborns, therefore, are especially susceptible to hypoglycemia when they are exposed to conditions that impair glucose homeostasis during the transition from intrauterine to extrauterine life.
 DEFINITION
DEFINITION
Hypoglycemia ought to be defined as any glucose concentration below the lower limit of the normal range of blood or plasma/serum glucose concentrations.5 This concentration, however, is uncertain, controversial, and variably defined. Early statistical evaluations in term infants defined hypoglycemia as a blood glucose concentration below 35 mg/dL, or a plasma glucose value below 40 mg/dL; even lower concentrations were used to define hypoglycemia in preterm infants. Such statistical definitions, however, have limited biological or clinical significance, as physiologically hypoglycemia is present when the concentration of glucose in the plasma produces glucose delivery rates that are inadequate to meet essential requirements for glucose utilization. Such requirements vary considerably among infants and over a broad range of glucose concentrations. Definitions of normal and hypoglycemic glucose concentrations also depend on postnatal feeding practices and when birth glucose concentrations are measured. Glucose concentrations are higher, for example, when feedings are initiated soon after birth than when feedings are delayed for several hours. Postnatally, the blood glucose concentration normally decreases to its lowest value between 1 and 3 hours after birth, followed by a progressive increase to greater than 50 to 60 mg/dL by 12 to 24 hours (Fig. 51-1).6
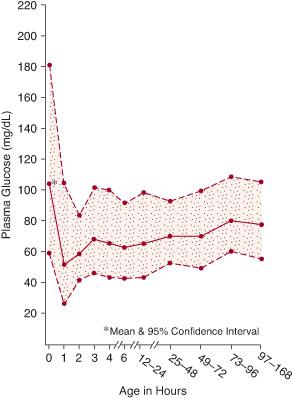
FIGURE 51-1. Plasma glucose concentrations during the first week of life in healthy, appropriate-for-gestational age term infants.
The absolute glucose concentration at or below which short- or long-term organ dysfunction occurs remains undefined, although animal studies suggest that concentrations below 20 mg/dL, when sustained over several hours, can result in brain injury.7 Without specific evidence to support an absolute threshhold value, no single value can be used to define physiological hypoglycemia. Recognizing this limitation, Rozance and Hay recently conducted a review of the neonatal hypoglycemia literature to determine the glucose concentrations and duration of hypoglycemia, and associated clinical signs that might indicate potential long-term injury (Table 51-1). Unfortunately, there are no sufficiently large, randomized controlled trials to establish the efficacy of any diagnostic and/or treatment approach, or any set of guidelines for the management of hypoglycemia (however defined) in the newborn. The key to preventing complications from glucose deficiency, therefore, is to focus less on numerical values of glucose concentration. Management should be directed toward identification of infants at risk, promotion of early and frequent feedings, normalization of glucose homeostasis, measurement of glucose concentrations early and frequently in infants at risk, and prompt treatment when glucose deficiency is marked and symptomatic.
Nevertheless, most clinicians today use higher values than previously accepted to define hypoglycemia. Several recent studies support this practice. Repeated blood glucose concentrations below 48 mg/dL in preterm infants were associated with adverse neurodevelopmental outcomes.8 Among a group of normal preterm infants, most achieved a serum glucose concentration greater than 50 mg/dL by 12 to 24 hours after birth.6 Human fetuses have a blood glucose concentration greater than 50 mg/dL during normal development.9 These observations indicate that blood glucose concentrations of 45 to 50 mg/dL (50–60 mg/dL plasma or serum) should represent the approximate lower limit for normal neonatal glucose concentrations.
Table 51-1. Necessary Conditions before Relating Long-Term Neurological Impairment to Neonatal Hypoglycemia
Blood or plasma glucose concentrations below 1 mmol/L (18 mg/dL). Such values definitely are abnormal, although if transient, there is no study in the literature confirming that they lead to permanent neurological injury. |
Persistence of such severely low glucose concentrations for prolonged periods (hours, probably > 2–3 hrs, rather than minutes, although there is no study in human neonates that defines this period). |
Early mild to moderate clinical signs (primarily those of increased adrenalin [epinephrine] activity), such as alternating central nervous system signs of jitteriness/tremulousness vs. stupor/lethargy or even a brief convulsion, that diminish or disappear with effective treatment that promptly restores the glucose concentration to the statistically normal range (> 45 mg/dL). |
More serious clinical signs that are prolonged (many hours or longer), including coma, seizures, respiratory depression and/or apnea with cyanosis, hypotonia or limpness, high-pitched cry, hypothermia, and poor feeding after initially feeding well. These are more refractory to short-term treatment. |
Concurrence of associated conditions, particularly persistent excessive insulin secretion and hyper-insulinemia with repeated episodes of acute, severe hypoglycemia with seizures and/or coma (although subclinical, often severe, hypoglycemic episodes occur in these conditions and might be just as injurious). |
Source: Rozance PJ, Hay WW Jr. Hypoglycemia in newborn infants: features associated with adverse outcomes. Biol Neonate. 2006; 90(2):74-86.
 DIAGNOSIS
DIAGNOSIS
The gold standard for measuring blood or plasma/serum glucose concentration is the hexokinase method used by most diagnostic laboratories. Colorimetric reagent strip methods are commonly used at the bedside to screen for hypoglycemia, but such measurements are often inaccurate, ranging from approximately 10% to 30% of the true value. Because of their relative inaccuracy, any low blood glucose concentration detected by one of these colorimetric methods must be confirmed by a standard laboratory method as soon as possible.
 INCIDENCE
INCIDENCE
The overall incidence of neonatal hypoglycemia has been estimated at 1 to 5 per 1000 live births; the incidence is higher in infants at increased risk of hypoglycemia.10 The reported incidence varies depending on the definition of hypoglycemia, the time after birth when the concentration of glucose is measured, and the method used to measure the blood, plasma, or serum glucose concentration.
 CLINICAL PRESENTATION
CLINICAL PRESENTATION
Hypoglycemia is classified as symptomatic or asymptomatic, terms that indicate the presence or absence of physical signs that accompany a low glucose concentration. Most signs are non-specific and result from disturbances in central nervous system function. The most common and least specific signs include tremulousness and irritability (often alternating with mild hypotonia), diminished arousal (often noted as stupor or lethargy as manifestations of mild changes in level of consciousness), and failure to eat after first eating well.5,11 These signs occur, however, with other common, similarly transient neonatal disorders, including hypocalcemia, subarachnoid hemorrhage, and early stages of sepsis. These signs usually correct quickly upon restoration of normal glucose concentrations. More severe and prolonged low glucose concentrations that do not respond as well to immediate treatment may be associated with abnormal respiratory patterns, such as respiratory depression or apnea leading to cyanosis; cardiovascular signs, such as tachycardia or bradycardia; and neurological signs, such as temperature instability, loss of consciousness or coma, limpness and hypotonia, and seizures.1 A diagnosis of hypoglycemia should be considered in any infant who exhibits any of these more serious signs, since untreated, severe, prolonged, and especially repeated hypoglycemia can impair the function of many organs, especially the brain.3 In those infants known to be at risk for hypoglycemia, careful observation and screening is required since intermittent, asymptomatic injury may occur that can lead to neurologic injury.
 ETIOLOGY
ETIOLOGY
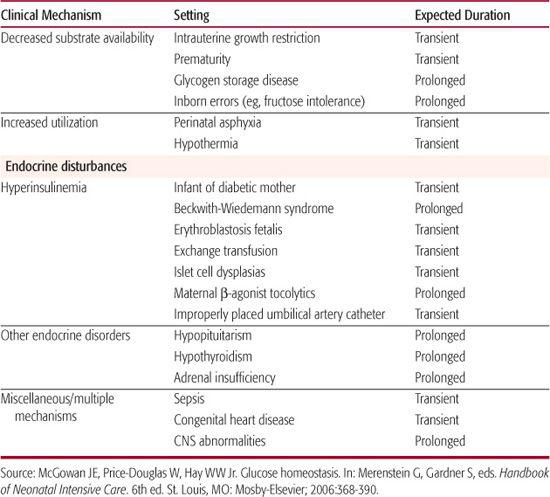
The most common cause of neonatal hypoglycemia is an imbalance of reduced glucose production and increased glucose utilization (Table 51-2).1,12 Decreased substrate availability is common among preterm and small-for-gestational-age infants with intrauterine growth restriction (see Chapter 47). 
Hypoglycemia that persists for more than 5 to 7 days most often results from several types of congenital hyperinsulinism.13 Although uncommon, these are serious metabolic disorders and are associated with a markedly increased risk of neurologic complications. Several of these disorders are genetic, including diffuse β-cell/islet hyperplasia or nesidioblastosis and an autosomal recessive form of congenital hyper-insulinism that has been linked to a defect in the sulfonylurea receptor or K+-ATP channel. A single mutation on the short arm of chromosome 11 in the Ashkenazi Jewish population also produces this defect. Cases in other ethnic groups have been associated with a number of other mutations in the same region. Mutations responsible for an autosomal dominant form of hyperinsulinism remain unknown. Unlike the recessive form, the autosomal dominant form of hyperinsulinism does not appear to result from abnormal sulfonylurea receptor function. A syndrome of congenital hyperinsulinemia and asymptomatic hyperammonemia associated with mutations in the glutamate dehydrogenase gene also has been described. Beckwith-Weidemann syndrome is associated with hyperplasia of multiple organs, including the pancreas, with increased insulin secretion. Rarely, hyperinsulinemia results from localized β-cell/islet adenomas within an otherwise normal pancreas.
Inborn errors of metabolism can limit availability of gluconeogenic precursors or the function of the enzymes required for hepatic glucose production. Metabolic defects that present with hypoglycemia include some forms of glycogen storage disease, galactosemia, fatty acid oxidation defects,14 carnitine deficiency, several of the amino acidemias, hereditary fructose intolerance (fructose-1,6-diphosphatase deficiency), and defects of other gluconeogenic enzymes. Endocrine disorders, such as hypopituitarism and adrenal failure, also lead to hypoglycemia because of inadequate hormonal responses, including inadequate growth hormone and adrenal corticosteroid secretion, resulting in failure to activate hepatic glucose production. These conditions are very rare and should be considered only after common etiologies are excluded. The approach to diagnosis of metabolic causes of hypoglycemia is discussed in Chapter 134, and endocrine causes of hypoglycemia are further described in Chapter 545.
 DIAGNOSIS
DIAGNOSIS
Risk factors for hypoglycemia should be identified, such as maternal diabetes or abnormal glucose tolerance, maternal administration of drugs associated with neonatal hypoglycemia, ultrasound evidence of intrauterine growth restriction, or preterm birth (eTable 51.1  ).1 Growth parameters should be plotted to establish if the infant is small or large for gestational age. Sepsis should be suspected if the infant has no apparent risk factors for hypoglycemia. If hypoglycemia persists for more than 1 week, hyperinsulinism, other endocrine disorders, and inborn errors of metabolism should be investigated, especially if the hypoglycemia is refractory to standard treatment. Hyperinsulinemia is diagnosed by demonstrating normal to high concentrations of insulin during episodes of hypoglycemia. Concentrations of insulin-like growth factor 1 binding protein (IGFBP-1) also are decreased in the presence of hyperinsulinemia. Serum and urine tests for specific metabolic and endocrine disorders, such as serum amino acid profiles and cortisol and growth hormone concentrations, also may help to determine the etiology of hypoglycemia.
).1 Growth parameters should be plotted to establish if the infant is small or large for gestational age. Sepsis should be suspected if the infant has no apparent risk factors for hypoglycemia. If hypoglycemia persists for more than 1 week, hyperinsulinism, other endocrine disorders, and inborn errors of metabolism should be investigated, especially if the hypoglycemia is refractory to standard treatment. Hyperinsulinemia is diagnosed by demonstrating normal to high concentrations of insulin during episodes of hypoglycemia. Concentrations of insulin-like growth factor 1 binding protein (IGFBP-1) also are decreased in the presence of hyperinsulinemia. Serum and urine tests for specific metabolic and endocrine disorders, such as serum amino acid profiles and cortisol and growth hormone concentrations, also may help to determine the etiology of hypoglycemia.
Table 51-2. Neonatal Hypoglycemia: Etiologies and Time Course
 PATHOPHYSIOLOGY
PATHOPHYSIOLOGY
Severe hypoglycemia in the newborn is associated with selective neuronal necrosis in multiple brain regions, including the superficial cortex, dentate gyrus, hippocampus, and cau-date-putamen. Brain injury appears to result from a number of processes that are initiated when blood glucose concentrations decrease.1,3 Energy failure leads to decreased cerebral electrical activity followed by neuronal cell membrane breakdown. Energy failure also prevents postsynaptic uptake of the principal neurotransmitter glutamate. Excess glutamate concentrations then activate NMDA (N-methyl D-aspartate) receptors in the neuronal membranes, which increases cellular entry and cytoplasmic concentrations of sodium and calcium to levels that cause osmotic swelling and acute neuronal necrosis. The high calcium concentrations also activate cellular phospholipases and proteases and prevent normal mitochondrial metabolism, which leads to increased toxic free radical formation. These processes disrupt synaptic transmission and eventually lead to delayed neuronal necrosis. Hypoglycemia also can exacerbate brain injury during periods of cerebral hypoxia.15 Hypoglycemia also abolishes hypoxic vasodilation of cerebral blood vessels, impairing compensatory mechanisms to improve oxygen delivery to the brain during periods of hypoxemia.
 MANAGEMENT
MANAGEMENT
Neonatal hypoglycemia must be corrected rapidly, and further episodes of hypoglycemia must be prevented by providing adequate substrate until normal glucose homeostasis is established (Fig. 51-2).1 Early enteral feeding usually is successful in treating mild hypoglycemia in asymptomatic infants. Feedings with 5% dextrose solution should be avoided because the dextrose is metabolized rapidly, and hypoglycemia frequently recurs. Human milk or standard infant formulas provide carbohydrate in the form of lactose without excessively stimulating insulin secretion. Milk also includes protein and fat, which provide a sustained supply of substrates for gluconeogenesis and alternative fuels. Fat intake also decreases cellular glucose uptake. Blood glucose concentrations should increase by 20 to 30 mg/dL within the first hour after a feeding of 30 to 60 mL of milk or formula.
Intravenous glucose infusion should be used when infants are symptomatic or are unable to tolerate enteral feedings and when hypoglycemia does not respond to enteral feeding. Intravenous glucose also should be used early after birth in those high-risk infants who are likely to experience severe disturbances in glucose homeostasis that are expected to last more than a few hours. Such infants include preterm infants, small-for-gestational-age infants with intrauterine growth restriction, infants of diabetic mothers, and infants who have underlying etiologies for hypoglycemia, such as sepsis, known or suspected inborn errors of metabolism, endocrine defects, or erythroblastosis. An initial rapid intravenous infusion of 200 mg/kg (2 mL/kg) of 10% dextrose solution should be followed by continuous infusion of 5 to 8 mg/min/kg of glucose—that is, the glucose utilization rate of a healthy term infant. The improved plasma glucose response to this regimen, compared to sole initiation of an 8 mg/kg/min infusion is shown in eFigure 51.1  . The blood glucose concentration should be measured approximately 30 minutes after the initial rapid infusion and then every 1 to 2 hours until it is stable within the normal range. If a subsequent value falls in the hypoglycemic range, the rapid infusion should be repeated, and the infusion rate increased by 10% to 15%.
. The blood glucose concentration should be measured approximately 30 minutes after the initial rapid infusion and then every 1 to 2 hours until it is stable within the normal range. If a subsequent value falls in the hypoglycemic range, the rapid infusion should be repeated, and the infusion rate increased by 10% to 15%.
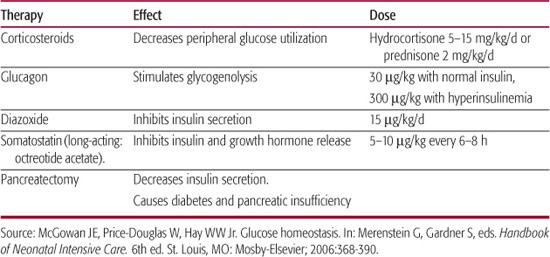
Infants with hyperinsulinemia often require as much as 12 to 15 mg/min/kg of intravenous glucose to maintain normoglycemia; these rates usually require a central venous catheter to allow infusion of dextrose concentrations greater than 12.5%. Infants requiring intravenous therapy for hypoglycemia should continue feedings as long as there is no evidence of feeding intolerance, such as abdominal distension, persistent and large residual volumes of milk in the stomach, or emesis. Providing some carbohydrate as galactose, one of the sugars that comprise lactose, is useful in infants of diabetic mothers and other infants with hyperinsulinism because pancreatic insulin response to galactose is less than the response to an equivalent amount of glucose. When a normal blood glucose concentration has been established, and the requirement for intravenous glucose has been stable for 12 to 24 hours, the infant can be weaned from this therapy by measuring preprandial blood glucose concentrations and decreasing the infusion rate by 10% to 20% each time the blood glucose is greater than 50 to 60 mg/dL. Failure to tolerate weaning from intravenous glucose indicates the presence of a pervasive disorder, such as a metabolic defect or idiopathic hyperinsulinemia.17 Several agents have been used to treat infants with refractory hypoglycemia, usually with coexisting hyperinsulinism (Table 51-3).1
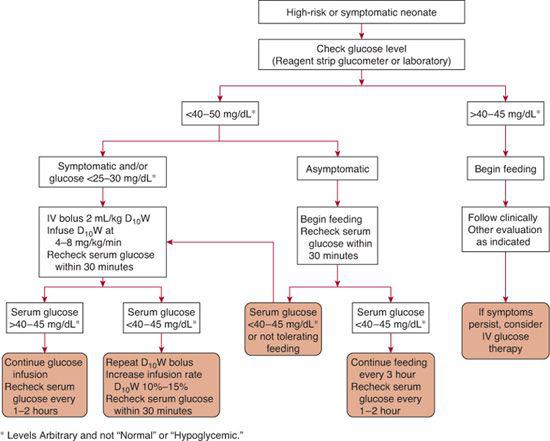
FIGURE 51-2. Algorithm for management of the neonate with hypoglycemia.
 OUTCOME
OUTCOME
Acute, severe hypoglycemia stimulates release of catecholamines and glucagon and subsequent lipolysis and glycogenolysis. The stress imposed by hypoglycemia can precipitate cardiorespira-tory instability in a previously ill infant. Long-term effects of severe neonatal hypoglycemia remain controversial. There is little evidence of long-term sequelae in term infants who have experienced relatively few, brief episodes of hypoglycemia. In preterm infants, however, repeated daily glucose concentrations below approximately 46 mg/dL have been associated with significantly lower mental and motor development and a higher incidence of cerebral palsy.18 Thus, mild to moderate hypoglycemia might affect outcome in high-risk infants, particularly those who cannot respond adequately to hypoglycemia, although such infants have many other confounding problems that independently or interactively lead to abnormal neurodevelopment.19
 HYPERGLYCEMIA
HYPERGLYCEMIA
Hyperglycemia is relatively common in infants who are born extremely preterm (< 26 weeks of gestation). Hyperglycemia is caused by excessive rates of intravenous glucose infusion in the presence of physiological and biochemical mechanisms that lead to excess glucose production, insulin resistance, and glucose intolerance.
Table 51-3. Adjunct Therapies for Persistent Neonatal Hypoglycemia
 DEFINITION
DEFINITION
Hyperglycemia is a blood glucose concentration greater than 120 to 125 mg/dL (plasma concentration > 145–150 mg/dL), regardless of gestational age, weight, or postnatal age.
 DIAGNOSIS
DIAGNOSIS
Affected neonates usually are extremely pre-term (< 26 weeks of gestation) and of extremely low birth weight (< 1000 g). They often are receiving relatively high rates of intravenous glucose. They usually are asymptomatic or have signs of associated disease processes. Recognizable findings associated with hyperglycemia include dehydration from osmotic diuresis, weight loss, fever, glycosuria, ketosis, and metabolic acidosis.20 The latter 3 signs are common among infants who have transient or permanent neonatal diabetes mellitus.
 EPIDEMIOLOGY
EPIDEMIOLOGY
The incidence of hyperglycemia is inversely related to birth weight in the preterm infant, ranging from about 2% in infants who weigh more than 2000 g to about 45% in those who weigh less than 1000 g, and up to 80% in infants weighing less than 750 g.
 ETIOLOGY
ETIOLOGY
Hyperglycemia occurs in about half of pre-term infants receiving continuous intravenous dextrose infusions that provide glucose at rates greater than 10 to 11 mg/min/kg and in nearly all infants at rates greater than 14 mg/min/kg.20-22 Even after dextrose infusion rates are decreased to treat hyperglycemia, infusion rates as low as 3 to 4 mg/min/kg have been associated with persistent hyper-glycemia, especially during prolonged periods of stress. Positive relationships also have been found between hyperglycemia and the severity of clinical problems in neonates, as estimated by lower Apgar scores, higher fractional concentrations of inspired oxygen, and worse respiratory distress scores. Intravenous lipid infusion also increases the incidence and degree of hyperglycemia, as increased plasma concentrations of free fatty acids decrease peripheral glucose utilization by competing with glucose for oxidation and by stimulating the activity of enzymes that specifically promote fatty acid oxidation.23 Fatty acids also promote glucose production by providing substrate and cofactors, such as nicotinamide adenine dinucleotide phosphate (NADPH), for gluconeogenesis, and they inhibit the suppressive effect of insulin on hepatic glucose production. Stress, as measured by increased plasma cortisol concentrations, also is an important risk factor for the development of hyperglycemia, occurring most frequently among infants receiving catecholamine infusions, or undergoing painful procedures such as surgery, venipuncture, vascular cutdowns, and endotracheal tube insertion without adequate analgesia or anesthesia. Narcotic treatment during and after surgery lowers the incidence of hyperglycemia.24 Hyperglycemia also occurs more commonly among preterm and small-for-gestational-age infants, who have increased plasma concentrations of counterregulatory (anti-insulin) hormones. Other more common causes of neonatal hyperglycemia include the use of medications such as theophylline and dexamethasone, and sepsis. Hyperglycemia has also been associated with prostaglandin E1 infusions.
Insulin-dependent diabetes mellitus, either transient or permanent, is an unusual but important cause of hyperglycemia in newborn infants.25,26 Neonatal diabetes mellitus usually presents early in postnatal life with weight loss, polyuria, dehydration, glycosuria, and hyperglycemia. It usually does not resolve for several weeks or even months and sometimes is permanent. Pathogenic mechanisms are not known. Hyperglycemia has been associated with specific chromosome deletions (e.g., 46,XXDq–on number 13) and with pancreatic agenesis.
 PATHOPHYSIOLOGY
PATHOPHYSIOLOGY
The principal mechanism responsible for hyperglycemia in preterm infants is intravenous infusion of dextrose at rates that exceed the capacity for glucose utilization.27 Glucose utilization is limited by relatively inadequate insulin secretion rates as well as by decreased peripheral insulin sensitivity (primarily in skeletal muscle) in infants who experience stress and relatively increased secretion rates of catecholamines, glucocorticoids, and glucagon. Even extremely low-birth-weight infants can have high rates of glucose production (4–8 mg/min/kg), primarily from gluconeogenesis, relatively soon after preterm birth.11
 COMPLICATIONS
COMPLICATIONS
There are few serious consequences of neonatal hyperglycemia, although this condition has been associated with a wide spectrum of sequelae that most likely are the result of pathologic conditions that also produce hyperglycemia.28,29 Another potential effect of aggressive glucose administration is steatosis, with associated impaired secretion of hepatic triglycerides. Steatosis rarely causes clinical signs in the neonate, but it can be detected by modest elevations of liver transaminases. Another complication of hyperglycemia is electrolyte imbalance in neonates who have glycosuria and increased sodium excretion. Recent evidence also links hyperglycemia in very preterm infants with an increased incidence of retinopathy of prematurity.31
 MANAGEMENT
MANAGEMENT
Treatment of hyperglycemia must include simultaneous treatment of underlying conditions. With modest hyperglycemia (plasma glucose concentration < 300 mg/dL), reducing the exogenous glucose (dextrose) infusion rate usually is sufficient to ameliorate or resolve the hyperglycemia. The rate of infusion should be decreased gradually by 1 to 2 mg/min/kg every 2 to 4 hours, with frequent measurement of glucose concentrations until normoglycemia is achieved.
Intravenous infusion of short-acting, regular insulin should be reserved for infants who have severe hyperglycemia (> 300 mg/dL) that persists despite reducing the glucose infusion rate to less than 3 to 4 mg/min/kg. These infants also may have increased serum lactate metabolic acidosis, hyperkalemia, and osmotic diuresis. Insulin is given by continuous intravenous infusion, beginning at 0.02 to 0.05 U/h/kg.32 Higher infusion rates are not necessary and increase the risks of hypokalemia and subsequent hypoglycemia. Hypokalemia can be prevented by addition of potassium to intravenous solutions during the insulin infusion. Small intravenous infusions of potassium (0.1 mEq/kg potassium as potassium chloride or potassium acetate) can be added every 1 to 2 hours if hypokalemia is significant and persistent. Dextrose solutions should be infused through a very secure intravenous line such as an umbilical venous catheter or a peripherally placed central venous line to prevent hypoglycemia that can occur when glucose infusion is interrupted. Blood glucose concentrations should be measured frequently, every 1 to 2 hours, or whenever signs of possible hypoglycemia develop.
Early intravenous amino acid infusion has been associated with decreased incidence and severity of hyperglycemia and hyperkalemia.33 The rationale for this approach is that certain amino acids, such as leucine, valine, isoleucine, glutamine, and arginine, are known insulin secretagogues and are important for normal growth and development of the pancreas and the pancreatic islets and β cells. Glutamine and leucine also may promote insulin action and the disposal of glucose in skeletal muscle. Enteral feeding—even a minimal enteral feeding regimen—also promotes the secretion of insulin by inducing gut production of enteroinsular hormones, also known as incretins, including gastric inhibitory polypeptide and pancreatic poly-peptide. These hormones increase insulin secretion by direct actions on the pancreatic cells. Thus, enteral feeding is appropriate to prevent or ameliorate hyperglycemia unless the infant is very ill or there are clear signs of feeding intolerance.
DISORDERS OF CALCIUM REGULATION
Disorders of calcium, phosphorus and magnesium metabolism are discussed in further detail in Chapter 542.
HYPOCALCEMIA
 DEFINITION
DEFINITION
Neonatal hypocalcemia is generally defined as a serum total calcium concentration of less than 2 mmol/L (< 8 mg/dL) in term infants and less than 1.75 mmol/L (< 7 mg/dL) in pre-term infants or an ionized calcium less than 0.75 to 1.1 mmol/L (< 3.0–4.4 mg/dL).34 Early-onset neonatal hypocalcemia typically is transient and occurs during the first 24 to 48 hours after birth; later-onset hypocalcemia usually occurs after the first week of life and commonly involves lasting pathology.35
 ETIOLOGY
ETIOLOGY
Early-onset hypocalcemia is most commonly associated with preterm birth, perinatal depression (from hypoxic-ischemic conditions), maternal insulin-dependant diabetes, gestational exposure to anticonvulsants, and maternal hyperparathyroidism. Previous experience was that transient hypocalcemia often occurred in approximately 30% of all preterm infants (up to 90% in those born extremely preterm), approximately 30% of infants who have an Apgar score below 5 at 1 minute of age, and approximately 10% to 20% of infants of insulin-dependant diabetic mothers. However, much lower rates are currently reported probably due to earlier postnatal administration of intravenous nutrient solutions that contain calcium and earlier onset of enteral feeding. Later-onset hypocalcemia is much less frequent and is most commonly associated with relatively high phosphate-containing diets, disturbed maternal vitamin D metabolism, intestinal malabsorption of calcium, hypomagnesemia, and hypoparathyroidism.
 PATHOPHYSIOLOGY
PATHOPHYSIOLOGY
Early neonatal hypocalcemia generally represents a transient failure of calciotropic hormone secretion in response to the loss of placental calcium supply at birth. Preterm infants often do not increase parathyroid hormone secretion, which can be aggravated by restricted intravenous and oral calcium intake, end-organ resistance to 1,25-dihydroxyvitamin D [1,25-(OH)2D], and increased serum calcitonin.36 Other contributing factors in infants exposed to hypoxic-ischemia conditions include an increased endogenous phosphate load, low neonatal glomerular filtration rate that limits phosphorous excretion, bicarbonate therapy, and an increased serum calcitonin concentration. Hypocalcemia in infants of insulin-dependant diabetic mothers appears to be related to magnesium insufficiency from renal losses and consequently to impaired fetal parathormone secretion. Formulas usually contain more phosphorus than human milk, and formula-fed infants often have lower serum ionized calcium and higher serum phosphorus in the first week of life compared to breast-fed infants.
Congenital hypoparathyroidism is the most significant cause of late-onset hypocalcemia that has to be treated early in life.37 Congenital hypoparathyroidism also occurs as part of the DiGeorge anomaly. Insufficiency of vitamin D arises from maternal vitamin D deficiency, sunshine deprivation coupled with insufficient dietary vitamin D intake, reduced production of vitamin D or its active metabolites caused by liver or renal disease, congenital deficiency of renal 1α-hydroxylase, and 1,25-(OH)2D resistance. Deficiency of vitamin D or its metabolites causes decreased intestinal calcium absorption and renal calcium reabsorption. Most infant vitamin D disturbances are not apparent until several months of age.38
Magnesium concentrations should be measured because it is necessary for parathyroid secretion and end-organ action.39
 CLINICAL PRESENTATION
CLINICAL PRESENTATION
Most cases of neonatal hypocalcemia are asymptomatic. When severe, the main clinical signs are jitteriness, tremors, twitching movements, and generalized or focal convulsions; infants also may be lethargic, feed poorly, vomit, and have abdominal distension.40 These findings are non-specific and are difficult to distinguish from those of hypoglycemia, polycythemia-hyperviscosity, or subarachnoid hemorrhage. Arrhythmias also may occur, but the characteristic prolongation of the electrocardiogram QT interval does not occur consistently in neonates with hypocalcemia. Frank convulsions are seen more commonly with late neonatal hypocalcemia (usually calcium < 6 mg/dL). The classical signs of peripheral hyperexcit-ability of motor nerves (carpopedal spasm and laryngospasm) are uncommon in newborn infants.
 DIAGNOSIS
DIAGNOSIS
The diagnosis of hypocalcemia is based on the determination of serum total and ionized calcium as well as phosphorus, magnesium, and glucose concentrations and serum pH. Electrocardiographic determination of prolonged corrected QT interval (> 0.40 sec) suggests hypocalcemia and may be useful to monitor response to calcium therapy.
Symptomatic hypocalcemia that is refractory to therapy may be caused by the less common causes of neonatal hypocalcemia, such as primary hypoparathyroidism, malabsorption, and disorders of vitamin D metabolism, which are identified by laboratory results. Elevated serum phosphorus concentration (> 8 mg/dL) indicates phosphorus loading (a clue to high dietary phosphorus intake), renal insufficiency, or hypoparathyroidism. Absence of a thymic shadow on chest radiograph suggests DiGeorge sequence. Hypercalciuria associated with hypocalcemia indicates deficient parathyroid hormone. Low serum 25-hydroxyvitamin D concentration (< 11 ng/mL) indicates vitamin D deficiency.
 MANAGEMENT
MANAGEMENT
Asymptomatic hypocalcemia is treated by providing intravenous calcium salts followed by formula or milk feeding providing 75 mg/kg/d of calcium. After normocalcemia is achieved, a stepwise reduction of intravenous calcium may help prevent rebound hypocalcemia: 75 mg/kg/d for the first day, half the dose the next day, half again, and discontinue. Infants of diabetic mothers often require as much as 100 to 200 mg/kg daily, and some require 2 to 3 times that amount. If infants can tolerate oral fluids, calcium gluconate can be given orally at the same doses (divided into 4 to 6 doses) after initial correction. Use of the intravenous form of calcium for enteral administration may cause less gut stimulation than syrup-based (high os-molar) preparations. Symptomatic hypocalcemia usually responds rapidly to intravenous calcium therapy, which helps confirm the validity of the diagnosis. When there are seizures, calcium can be replaced rapidly with 1 to 2 mL of 10% calcium gluconate per kg (9–18 mg elemental calcium/kg) given intravenously over 5 to 10 minutes; heart rate should be measured continuously during the infusion because of bradycardia, for which the infusion should be stopped temporarily. Calcium can cause considerable tissue injury with extravasation from the vein, requiring careful intravenous insertion site monitoring. Vitamin D metabolites are not recommended for treatment of early hypocalcemia because of their variable responses and potential side effects.
Most cases of neonatal hypocalcemia are transient and need only 2 to 3 days of treatment, but such treatment is important to prevent adverse cardiovascular and central nervous system complications of low serum calcium concentrations. Calcium supplementation is usually required for long periods in the case of hypocalcemia caused by malabsorption or hypoparathyroidism.41 If hypocalcemia is associated with hypomagnesemia (serum magnesium concentration below 0.6 mmol/L or 1.5 mg/dL), magnesium sulfate 50% solution (500 mg or 4 mEq/mL), 0.1 to 0.2 mL/kg intravenously or intramuscularly, providing 50 to 100 mg/kg (which also may cause local tissue necrosis if extravasated), should be given and repeated after 12 to 24 hours. Serum magnesium should be obtained before each dose, as 1 or 2 doses may resolve transient hypomagnesemia. This treatment, however, corrects the hypomagnesemia and is necessary to also correct the hypocalcemia, but prophylactic magnesium treatment of infants of diabetic mothers does not prevent their hypocalcemia.
Late hypocalcemia usually is symptomatic and requires treatment. The goals of therapy are to reduce the phosphorus load and to increase calcium absorption by using feedings with a calcium/phosphorus ratio greater than or equal to 4:1. This can be accomplished by the use of low-phosphorus feedings, such as human milk or low-phosphorus formula, in conjunction with an oral calcium supplement. Phosphate binders generally are not necessary. Hypoparathyroidism requires therapy with vitamin D or preferably one of its metabolites: 1,25-dihydroxyvitamin D (or 1α-hydroxyvitamin D3, a synthetic analog) and life-long calcium supplementation (see Chapter 542).
 PREVENTION
PREVENTION
Early neonatal hypocalcemia can be prevented by oral or intravenous calcium supplementation (75 mg elemental calcium/kg/d). Use of continuous calcium infusion by central catheter to maintain a total calcium higher than 8.0 mg/dL and an ionized calcium level higher than 4.0 mg/dL may be helpful to prevent hypocalcemia in sick newborns who exhibit cardiovascular compromise and require cardiotonic drugs or pressure support. Maintenance of normal maternal vitamin D status with exogenous vitamin D supplement may be helpful for maintaining adequate amounts of fetal vitamin D, which, in turn, may prevent late hypocalcemia in some neonates. In addition to the measures just described, judicious use of bicarbonate administration and avoidance of respiratory alkalosis both reduce the risk of developing symptomatic hypocalcemia in ill infants.
Regular follow-up monitoring of serum calcium concentration and appropriate monitoring of underlying disease (eg, parathyroid hormone concentration) are necessary in those infants who continue to have hypocalcemia due to variable forms of hypoparathyroidism that may last for several years, even into adolescence.
HYPERCALCEMIA
 DEFINITION
DEFINITION
Neonatal hypercalcemia is defined as a serum total calcium concentration greater than 2.75 mmol/L (11 mg/dL) or an ionized calcium concentration greater than 1.4 mmol/L (5.6 mg/dL).42 Pathological hypercalcemia includes increased ionized and total calcium, although increased total calcium may occur without increased ionized calcium.
 ETIOLOGY
ETIOLOGY
Hypercalcemia is not common but occurs relatively more frequently in preterm infants.43 The most common cause of hypercalcemia is a relative deficiency in phosphate supply and hypophosphatemia caused by use of parenteral nutrition with insufficient phosphate administration (with or without excessive calcium).35 Hypercalcemia also results from excessive administration of calcium (eg, overzealous use of calcium to treat hypocalcemia or to prevent it during exchange transfusion) or vitamin D. Hypercalcemia of the mother and the neonate may result from chronic maternal exposure to excess vitamin D or its metabolites during treatment of maternal hypocalcemia or by maternal self-medication. Chronic diuretic therapy with thiazides during pregnancy also can cause maternal and fetal hypercalcemia and subsequently neonatal hypercalcemia.
Much rarer causes of hypercalcemia in the newborn have a metabolic or familial basis, such as primary hyperparathyroidism, familial hypocalciuric hypercalcemia, hypercalcemia associated with subcutaneous fat necrosis, idiopathic infantile hypercalcemia (which may be part of Williams syndrome), severe infantile hypophosphatasia, and Bartter syndrome variant.44
 PATHOPHYSIOLOGY
PATHOPHYSIOLOGY
Normally, an increase in serum calcium inhibits parathyroid hormone and 1,25-dihydroxyvitamin D synthesis and thereby prevents or reduces hypercalcemia by decreasing calcium mobilization from bone, absorption from intestine, and reabsorption from kidney.45 A sustained elevation in serum calcium concentration implies an inappropriately increased calcium efflux from one of these pools into the extracellular fluid.
Hypophosphatemia can cause elevated circulating 1,25-(OH)2D with attendant increased intestinal absorption of calcium and increased bone resorption; calcium cannot be deposited in bone in the absence of phosphate and thereby contributes to hypercalcemia. Pathological conditions associated with parathyroid hormone or vitamin D overactivity that lead to hypercalcemia include increased bone turnover, intestinal calcium absorption, and renal calcium absorption.
 CLINICAL PRESENTATION
CLINICAL PRESENTATION
Hypercalcemia may be asymptomatic, or the infants may have mild to serious clinical signs. Mild conditions include nonspecific signs such as lethargy, irritability, poor feeding, vomiting, constipation, polyuria, dehydration, feeding difficulties, and poor growth. More serious signs include seizures, hypertension, respiratory distress (from hypotonia, demineralization, and deformation of the rib cage), and nephrocalcinosis. Long-standing hypercalcemia can lead to metastatic calcifications such as nephrocalcinosis.
 DIAGNOSIS
DIAGNOSIS
An increased serum calcium concentration confirms the diagnosis. A maternal dietary and drug history (eg, excessive vitamin A or D, thiazides) or history of possible calcium-phosphorus imbalance or polyhydramnios during pregnancy, or a family history of disturbed calcium metabolism, should prompt further evaluation. Very elevated serum calcium level (> 15 mg/dL) usually indicates primary hyperparathyroidism or, in very low-birth-weight infants, phosphate depletion. Further evaluation should include serum phosphorus concentration, percentage renal tubular phosphorus reabsorption, serum parathyroid hormone concentration, urinary calcium/urinary creatinine ratio (Uca/Ucr), serum alkaline phosphatase concentration, and serum 25-hydroxyvitamin D concentration. Bone x-ray films will identify demineralization and/or osteolytic lesions or osteosclerotic lesions (occasionally with vitamin D excess) consistent with the etiology of the hypercalcemia.
 MANAGEMENT
MANAGEMENT
Therapy of neonatal hypercalcemia consists of correction of specific underlying causes and removal of iatrogenic or external causes, for example, surgical removal of hyperparathyroid glands, stopping of excessive vitamin D intake, and phosphate (and calcium) supplementation of human milk given to pre-term infants.46 In mild cases in which patients are asymptomatic, maintenance of hydration may suffice. For a moderately to severely hypercalcemic infant, prompt investigation and therapy must be instituted with the following goals: correction of dehydration, enhancement of renal excretion of calcium, inhibition of intestinal absorption or bone resorption, restriction of dietary calcium intake, and treatment of the underlying disorder.
For short-term treatment of acute symptomatic hypercalcemic episodes (or serum calcium > 14 mg/dL), expansion of the extracellular fluid compartment with 10 to 20 mL/kg of 0.9% sodium chloride intravenously, followed by an intravenous injection of a potent loop diuretic such as 1 mg/kg of furosemide every 6 to 8 hours, may increase urinary calcium excretion and decrease serum calcium; electrolyte balance must be monitored during this treatment. In hypercalcemic patients with low serum phosphorus concentrations, phosphate supplements of 0.5 to 1.0 mmol (16–31 mg) of elemental phosphorus per kilogram per day in divided oral doses may normalize the serum phosphorus concentration and lower serum calcium concentration; parenteral phosphate, however, should be avoided in severely hypercalcemic patients (serum total calcium > 12 mg/dL) unless hypophosphatemia is severe (< 1.5 mg/dL) because extraskeletal calcification theoretically may occur. Calcitonin, glucocorticoids, bisphosphonate, and dialysis also have been used for severe hypercalcemia.47 For severe and unremitting hypercalcemia, either hemodialysis if the infant is hemodynamically stable or peritoneal dialysis may be helpful. Virtually all cases of primary hyperparathyroidism require subtotal or total parathyroidectomy, because the hypercalcemia may be life threatening and does not respond to medical management.48 The need for treatment should be reassessed at regular intervals because some instances of neonatal hypercalcemia may resolve spontaneously.
COMMON NEONATAL FLUID, ELECTROLYTE, AND ACID-BASE DISORDERS
HYPERNATREMIA
 DEFINITION
DEFINITION
Hypernatremia is defined as a serum sodium concentration greater than 150 mEq/L.49
 INCIDENCE AND EPIDEMIOLOGY
INCIDENCE AND EPIDEMIOLOGY
Hypernatremia is a common problem in pre-term infants, particularly those born before 28 weeks gestation and of extremely low birth weight (< 1000 g). The frequency of severe hypernatremia has diminished in recent years as clinicians have learned to limit sodium intake and have prevented dehydration by improved fluid management.50
 ETIOLOGY AND PATHOPHYSIOLOGY
ETIOLOGY AND PATHOPHYSIOLOGY
The most common cause of hypernatremia is dehydration, usually caused by too little “free water” administration. It is necessary to include some sodium and dextrose (usually at least 4%) in administered intravenous solution to assure that their osmolality is close to that of plasma to prevent hemolysis in the blood. The added sodium contributes to the hypernatremia, and the added dextrose contributes to the hyperglycemia. Excessive evaporative water loss across the very underdeveloped skin of extremely premature infants also contributes to dehydration and hypernatremia. Evaporation is aggravated by radiant warmers, high environmental temperatures, increased body temperatures, low ambient or inspired humidity, congenital skin defects (eg, omphalocele), and phototherapy. Other important contributing factors to hypernatremia include the often very high rates of urinary water loss due to poor concentrating capacity of the underdeveloped renal medulla (see Chapter 465).51,52 Hypernatremia in preterm infants also is frequently caused by injudicious use of sodium-containing solutions. The most common offender is the controversial use of normal saline bolus infusions to treat low blood pressure, which is often not necessary. Hypernatremia results when fluids are subsequently restricted to treat fluid overload. These same infants also may receive bolus infusions of sodium bicarbonate to treat metabolic acidosis, although this practice should be discouraged because its risks usually outweigh its presumed benefits. Sodium-containing medications often are overlooked as contributors to hypernatremia. Uncommonly, congenital or acquired reduction in antidiuretic hormone (diabetes insipidus) can lead to excessive, relatively sodium-free urinary water loss (see Chapter 525).
Sodium is the principal cation of extracellular fluid, including the plasma. Hypernatremia from dehydration causes cellular dehydration in response, and this can occur in any organ and tissue in the body, particularly the brain. Hypernatremia accompanied by increased fluid, such as from repeated normal saline bolus infusions, produces hypervolemia and the risk of patent ductus arteriosus, particularly if the infant has reduced urine flow rate and has not yet mounted a natriuretic hormone response (this is more common in the first 2–3 days of life).53
 CLINICAL PRESENTATION
CLINICAL PRESENTATION
Clinical signs of hypernatremia are rare except for late-occurring seizures with very severe hypernatremia, which usually indicates intracranial hemorrhage.
Mild to moderate hypernatremia usually is well tolerated, but it can contribute to patency of the ductus arteriosus, particularly when the hypernatremia is produced by normal saline infusions or high water infusion rates leading to transient hypervolemia. More severe hypernatremia leads to osmotic shifts in capillaries and cells in the central nervous system, which can rupture and produce intracerebral hemorrhage and neuronal cell necrosis. The incidence of intracranial hemorrhage in pre-term infants is directly related to sodium intake as well as, secondarily, to serum sodium concentration.54
 DIAGNOSIS AND MANAGEMENT
DIAGNOSIS AND MANAGEMENT
Hypernatremia is diagnosed by relatively frequent measurements of serum electrolyte concentrations, particularly in those very small infants whose skin appears extremely immature and those receiving low fluid and high sodium intakes.
Management should include use of plastic body bags or other means to decrease evaporative water loss, frequent measurements of weight and urinary flow rate to adjust fluid intake to prevent dehydration, and more judicious use of sodium chloride and avoidance of sodium bicarbonate bolus infusions. Treatment is directed toward correcting the cause.51,55 If hyperglycemia is a problem and requires reduction of intravenous fluid rates, sterile water can be infused into the stomach via nasogastric or orogastric tubes.56 Serum sodium should be reduced slowly to prevent osmotic swelling in cells, particularly in the central nervous system, which can lead to cell damage and seizures, aiming to correct no more than 10 mEq/L of serum sodium concentration over 24 to 36 hours (see Chapter 466).57,58 Extremely severe hypernatremia, particularly if the infant has central nervous system signs such as seizures and intracerebral hemorrhage, can be treated with whole blood exchange transfusion.
 OUTCOME
OUTCOME
The occurrence of cerebral palsy correlates strongly with a history of newborn hypernatremia, particularly when associated with intracranial hemorrhage and neuronal necrosis.59
HYPONATREMIA
 DEFINITION
DEFINITION
Hyponatremia is defined as a serum sodium concentration less than 130 mEq/L.
 INCIDENCE AND EPIDEMIOLOGY
INCIDENCE AND EPIDEMIOLOGY
Hyponatremia is common in extremely small preterm infants, perhaps even more so than hypernatremia. Usually, these infants are treated in the first 2 to 3 days of life with high intravenous fluid infusion rates to prevent dehydration at the same time that they have reduced glomerular filtration rates and urinary flow rates associated with pulmonary disorders.49 Use of body bags, 100% humidification of incubator air and ventilator or continuous positive airway pressure circuits, and other measures to diminish evaporation contribute to overhydration. Late hyponatremia also is common, usually as a result of diuretic use.58
 ETIOLOGY AND PATHOPHYSIOLOGY
ETIOLOGY AND PATHOPHYSIOLOGY
The most common cause of hyponatremia in preterm infants is overhydration from excessive administration of electrolyte-free intravenous solutions.60-62 This also can occur at birth when the mother receives high intravenous infusion rates during labor or cesarean section; water diffuses across the placenta more rapidly than do solutes, and because of active transport of amino acids and calcium, fetal plasma can have a higher osmolality than the mother’s. Decreased effective blood volume also prevents the suppression of antidiuretic hormone (vasopressin) secretion, common with perinatal depression secondary to hypoxic-ischemic conditions, intracranial hemorrhage, sepsis, respiratory distress, intrathoracic air leak syndromes, and medications such as morphine, barbiturates, or carbamazepine. Low glomerular filtration rates at early gestational ages also limits high urine flow rates in response to fluid boluses, particularly in the first 2 to 3 days of life (see Chapter 465). Further dilution of serum sodium often is augmented by coincident hyper-glycemia, which osmotically pulls water from intracellular and extravascular extracellular fluid spaces. Later hyponatremia occurs frequently as a result of renal loss of sodium from diuretic therapy, particularly the potent loop diuretics such as furosemide. The syndrome of inappropriate antidiuretic hormone secretion also can produce hyponatremia, which is suspected clinically when decreased serum sodium concentrations and urine output occur simultaneously, usually in response to a sudden, often catastrophic change in central nervous system and cardiovascular status, such as with acute, severe intracranial hemorrhage.58 Suboptimal sodium intake at any stage contributes to hyponatremia, as does urinary sodium loss once the very pre-term infant enters its natural diuretic phase after the first 2 to 3 days of life. Late hyponatremia has been noted with exclusive breast milk feedings without supplements of milk fortifiers that include salts and protein and with certain low-sodium formulas.
Low sodium concentrations in the plasma and extravascular/extracellular fluid prompt increases in urine flow rate and sodium retention, but sodium retention often does not keep up with water loss, leading to further sodium wasting. Water from the extracellular fluid also diffuses into cells where the normally high potassium concentration is relatively maintained, leading to osmotic swelling49 (in the brain, this can lead to increased intracranial pressure and seizures from disturbed action potential development and propagation, although these are late complications of relatively severe hyponatremia).
 CLINICAL PRESENTATION
CLINICAL PRESENTATION
Hyponatremia is usually asymptomatic because of chronic rather than acute development of sodium imbalance, but a late clinical sign is seizures. Infants fed unsupplemented breast milk or low-sodium formulas often develop peripheral edema with hyponatremia that sometimes is aggravated by hypoproteinemia from low protein in the diet.
Severe and persistent hyponatremia is associated with pathologic conditions in the central nervous system (seizures), lung (patent ductus arteriosus, pulmonary edema), and acid-base balance (hypochloremic alkalosis from diuretic therapy, often worsened by bicarbonate generation from retained carbon dioxide in infants with chronic lung disease), as well as poor growth that often is accompanied by peripheral edema.
 DIAGNOSIS AND MANAGEMENT
DIAGNOSIS AND MANAGEMENT
Diagnostic criteria include (1) low serum sodium, (2) continued urine sodium loss, (3) urine osmolality greater than plasma, and (4) normal adrenal and renal function.
Hyponatremia is identified by frequent measurements of serum electrolyte concentrations. Management is by water restriction until diuresis follows and is directed toward the etiology.51 Diuretics that often are used to reduce pulmonary edema, particularly in infants with chronic lung disease, only aggravate the hyponatremia and promote hypochloremic alkalosis.
 OUTCOME
OUTCOME
Infants with hyponatremia may grow more slowly, although this probably is related to associated nutritional and acid-base disturbances, and they more commonly develop cerebral palsy, although this has not been demonstrated to be an independent risk factor separate from associated pathologies (eg, intracranial hemorrhage) and preterm birth itself.59
HYPERKALEMIA
 DEFINITION
DEFINITION
Hyperkalemia is defined as a serum potassium concentration greater than 7 mEq/L, but the rate of rise is more important than the exact concentration.
 INCIDENCE AND EPIDEMIOLOGY
INCIDENCE AND EPIDEMIOLOGY
Hyperkalemia occurs frequently in very pre-term infants, usually in the first few days of life in association with other serious conditions and aggressive forms of treatment. The most common cause of laboratory reports of hyper-kalemia is cellular damage and leakage of potassium into blood collected by traumatic heel stick; usually, this is noted on the laboratory report as a “hemolyzed specimen”; lack of specific electrocardiographic changes and absence of other specific clinical signs tempers need to repeat such samples.63
 ETIOLOGY AND PATHOPHYSIOLOGY
ETIOLOGY AND PATHOPHYSIOLOGY
Causes of hyperkalemia include (1) acidosis with or without tissue destruction, (2) renal failure, particularly with continued potassium treatment, and (3) adrenal insufficiency (relatively uncommon). Hyperkalemia often occurs with hyperglycemia in very preterm infants receiving high dextrose infusion rates, and some even refer to this problem as the “hyperglycemia-hyperkalemia syndrome.”
Potassium is the main cation of intracellular space and contributes to polarization of the cell membrane. High extracellular fluid potassium concentrations, therefore, tend to depolarize the cell membrane and contribute to reduced action potentials in nerves and conductive cells in skeletal, cardiac, and smooth (eg, gut) myocytes.63,64
 CLINICAL PRESENTATION
CLINICAL PRESENTATION
Clinical signs include muscle weakness (hypotonia), ileus, and a variety of cardiac dysrhythmias. Electrocardiographic changes include prolonged of PR interval, peaked of T waves, widened QRS duration, and subsequently a sine-wave pattern of the QRS-T waves and ventricular fibrillation.63
When severe, hyperkalemia can produce life-threatening cardiac dysrhythmias, including asystole. Ileus can contribute to feeding intolerance. Muscle weakness in the diaphragm and thoracic muscles can aggravate other causes of respiratory failure.
 DIAGNOSIS AND MANAGEMENT
DIAGNOSIS AND MANAGEMENT
Hyperkalemia is determined by frequent measurements of serum electrolyte concentrations; its severity, along with clinical and electrocardiographic signs, determines the need and urgency of treatment.
Serum electrolytes should be measured frequently. Management is directed toward the causes and nonspecific treatment, depending on severity of the hyperkalemia.65 Nonpotassium-containing volume expanders can acutely lower high serum potassium concentrations.66
When hyperkalemia is severe, the following actions are urgent:58
• Stop all potassium administration.
• Infuse 100 to 200 mg/kg of calcium gluco-nate to lower the cell membrane threshold (this is transient but may be lifesaving).
• Infuse sodium bicarbonate, 1 to 2 mEq/kg intravenously, over at least 30 minutes. This transient therapy enhances intracellular sodium and hydrogen exchange for potassium and is particularly useful when the hyperkalemia is associated with acidosis. If hyperkalemia is associated with acute renal failure, the volume of fluid necessary to deliver the sodium bicarbonate may be excessive.
• Administer 1 g/kg cation exchange resin (sodium polystyrene-sulfonate [Kayexalate]) as an oral or rectal solution. Little experience has been reported in neonates, and technical problems of retention can be substantial. Furthermore, this may not be an option if the infant is restricted to nothing by mouth or has an injured gastrointestinal tract. When this resin is used, sodium from the resin is exchanged for serum potassium, which may result in hypernatremia, requiring frequent serial electrolyte measurements and increased fluid administration, often with diuretics.
• An insulin infusion, given simultaneously with a dextrose infusion, can help shift potassium intracellularly. All of the complications of insulin infusion (see section on treatment of severe hyperglycemia) can occur with this treatment for severe hyper-kalemia.
• Perform peritoneal dialysis; frequently, sodium bicarbonate must be added to the dialysate to prevent acidosis. Peritoneal dialysis takes time to set up. It may be technically impractical in very preterm infants.
HYPOKALEMIA
 DEFINITION
DEFINITION
Hypokalemia is defined by a serum potassium concentration less than 3.5 mEq/L.
 INCIDENCE AND EPIDEMIOLOGY
INCIDENCE AND EPIDEMIOLOGY
Hypokalemia is most common in very pre-term infants during diuretic therapy.
 ETIOLOGY AND PATHOPHYSIOLOGY
ETIOLOGY AND PATHOPHYSIOLOGY
The most common causes of hypokalemia are (1) increased gastrointestinal losses from an ostomy or drainage from a nasogastric tube when there is lower intestinal obstruction, and (2) renal losses, common in diuretic therapy.58,63 Hypokalemia also can occur when insulin and glucose infusions are used to treat severe hyperglycemia.65
About 90% of total potassium is intracellular. Loss of intracellular potassium depolarizes cells, leading to cellular dehydration and diminished action potentials in nerves, cardiomyocytes (particularly those involved in contractile conduction), and gut myocytes (leading to ileus).
 CLINICAL PRESENTATION AND COMPLICATIONS
CLINICAL PRESENTATION AND COMPLICATIONS
Clinical signs of hypokalemia are related to muscle weakness (generalized hypotonia and reduced respiratory effort), cardiac dysrhythmias (abnormal conductions, but most seriously, asystole and cardiac arrest), and diminished gut peristalsis (producing ileus and feeding intolerance).63,64
 DIAGNOSIS AND MANAGEMENT
DIAGNOSIS AND MANAGEMENT
Serum electrolyte concentrations should be measured frequently to document the disorder. Early electrocardiographic changes include decreased T wave amplitude and ST-segment depression.58
Management is directed toward the causes, such as correction of metabolic acidosis and ostomy and renal losses. Low serum potassium always implies significant intracellular depletion, but intracellular potassium can be low with normal serum potassium. When intravenous treatment is used, intravenous solutions usually should not contain more than 40 mEq/L of potassium, and the treatment should be over hours, not by bolus (except for life-threatening cardiac dysrhythmias). Diuretic-induced hypokalemia can be minimized by choosing a potassium-sparing diuretic such as spironolactone and increasing potassium salt supplements of milk or formula.58,66
METABOLIC ACIDOSIS
 DEFINITION
DEFINITION
Metabolic acidosis is a decrease in pH due to increased acid (H+ hydrogen ion or proton) production greater than any coincident increase in bicarbonate or decrease in PCO2 or a loss of bicarbonate greater than any coincident decrease in PCO2 or decrease in acid production.
 INCIDENCE AND EPIDEMIOLOGY
INCIDENCE AND EPIDEMIOLOGY
Metabolic acidosis is common in preterm infants due to hypoxic-ischemic conditions that produce lactic acidosis, as well as bicarbonate loss in the urine or plasma dilution by rapid fluid expansion of the blood volume and extracellular fluid.67,68
 ETIOLOGY AND PATHOPHYSIOLOGY
ETIOLOGY AND PATHOPHYSIOLOGY
Increased acid production from hypoxia-ischemia is common at birth from problems during labor and delivery or following catastrophic events such as intracranial hemorrhage, acute and severe endotoxemia with hypotension from sepsis, or blood volume loss.69 Acute renal failure also can decrease acid excretion. Bicarbonate loss in the diuresis phase after birth in very preterm infants also is common and often aggravated by dilutional acidosis from excessive volume administration (saline boluses to treat low blood pressure), high fluid infusion rates and low urine flow rates.70
Metabolic acidosis is categorized as increased anion gap or normal anion gap acidosis.63Increased anion gap acidosis occurs with the addition of acid to the blood and extracellular fluid, such as with lactic acidosis, renal failure, and metabolic disorders. The serum chloride concentration is normal in these disorders. Normal anion gap acidosis occurs with bicarbonate loss in the urine (proximal renal tubular acidosis), ostomy drainage, the inability to excrete acid due to defects in distal nephron function (distal renal tubular acidosis), or administration of Cl-containing compounds (eg, arginine HCl, HCl, CaCl2, MgCl2, NH4Cl hyperalimentation, high-protein formula). The serum chloride is increased in these disorders. Mild to moderate, chronic metabolic acidosis occurs in infants fed too much protein (> 4–5 g/kg/d) and may contribute to growth failure.71-73 Acute and relatively severe metabolic acidosis has been associated with decreased cardiac contractile performance and increased pulmonary vascular tone (pulmonary hypertension), although the degree that such conditions are due just to increased acid in the blood is controversial and probably not as marked as previously assumed.
 CLINICAL PRESENTATION
CLINICAL PRESENTATION
Acute metabolic acidosis usually is associated with clinical conditions that produce it. Chronic metabolic acidosis often is accompanied by growth failure.71
Complications of metabolic acidosis usually are those of underlying causes or inappropriate treatments.
 DIAGNOSIS AND MANAGEMENT
DIAGNOSIS AND MANAGEMENT
Metabolic acidosis is measured by blood gas analysis. It should be expected with acute abnormalities that include hypoxia, ischemia, hypovolemia, hypotension, and aggressive intravenous fluid treatment. It should be anticipated and looked for with more chronic conditions such as high urinary flow rates, excessive ostomy or gastric drainage, and poor growth.74
Acid production should be diminished by correcting the underlying pathophysiology. Acute intravenous sodium bicarbonate bolus infusion has been commonly used to treat severe and persistent metabolic acidosis, but this treatment generally is unproven and quite risky.67,68,75 Bicarbonate infusions produce blood volume overload, intracranial hemorrhage, hypernatremia, respiratory acidosis, decreased capillary oxygen exchange from increased hemoglobin-oxygen affinity, and a paradoxical intracellular acidosis as carbon dioxide, produced when the bicarbonate reacts with water via carbonic anhydrase, diffuses into cells. This treatment should be reserved for severely unstable and acidotic infants when other measures fail.76 Infants should be intubated and ventilated or spontaneously breathing sufficiently to lower their PCO2 easily. Infants with dilutional acidosis should have their fluid balance corrected. Chronic metabolic acidosis from bicarbonate loss responds well to daily addition of bicarbonate to feedings.
 OUTCOME
OUTCOME
Chronic metabolic acidosis produces reversible growth failure. Outcomes from acute, severe metabolic acidosis are usually due to its underlying causes or overly aggressive treatment with sodium bicarbonate bolus infusions.
METABOLIC ALKALOSIS
 DEFINITION
DEFINITION
Metabolic alkalosis is an increase in pH due to loss of acid or gain of bicarbonate without sufficient increase in PCO2.
 INCIDENCE AND EPIDEMIOLOGY
INCIDENCE AND EPIDEMIOLOGY
Metabolic alkalosis occurs frequently in newborns but is common to only a few specific conditions.
 ETIOLOGY AND PATHOPHYSIOLOGY
ETIOLOGY AND PATHOPHYSIOLOGY
Three basic mechanisms contribute to metabolic alkalosis: loss of acid such as hydrochloric acid from vomiting or intestinal obstruction and drainage; excessive treatment with or ingestion of base, such as alkali bolus infusions during resuscitations; and contraction of the extracellular fluid from dehydration (contraction alkalosis) or from loss of fluids containing more chloride than bicarbonate (as occurs with chronic diuretic treatment).63 Common causes of metabolic alkalosis in neonates include acid loss from vomiting with pyloric stenosis or duodenal stenosis/atresia or other high intestinal obstructions) or gastric drainage following intestinal surgery or with persistent ileus.70 Potassium depletion promotes metabolic alkalosis by stimulating renal ammonia genesis and inhibiting movement of hydrogen ions out of cells. Chloride depletion or chronic respiratory acidosis also maintains a metabolic alkalosis.74
Bicarbonate usually can be excreted by the kidney, except with dehydration and reduced glomerulus filtration rate that diminishes urine flow rate and distal tubule chloride-bicarbonate exchange.63,74 This condition also promotes proximal bicarbonate resorption that occurs readily with sodium resorption promoted by aldosterone secreted in response to low glomerulus filtration rate or blood volume as well as the hyponatremia.
 CLINICAL PRESENTATION
CLINICAL PRESENTATION
Metabolic alkalosis usually is asymptomatic but should be suspected in infants with high urine flow rates, ostomy and gastric drainage, and those treated with alkali and/or hyperventilation.
Complications of metabolic alkalosis usually are those of associated conditions.
 DIAGNOSIS AND MANAGEMENT
DIAGNOSIS AND MANAGEMENT
Metabolic alkalosis is diagnosed by blood gas measurements.
Underlying pathophysiology must be corrected. Chronic and marked contraction alkalosis can occur with intracellular potassium deficiency that often is more severe than indicated by the serum potassium concentration.
 OUTCOME
OUTCOME
Chronic alkalosis has been associated with sensorineural hearing loss that is distinct from commonly associated loop diuretic ototoxicity. Neurodevelopmental outcomes appear to be worse with acute, severe alkalosis produced by hyperventilation (respiratory alkalosis) and alkali treatment; the patho-physiology causing this adverse outcome is not clearly defined.75
REFERENCES
See references on DVD.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


