Hemoglobinopathies
Charles T. Quinn
Hemoglobin (Hb) is the oxygen-carrying protein within red blood cells (RBCs). It is composed of four globular protein subunits, called globins, and four oxygen-binding heme groups, which are attached to each globin. The two main types of globins are the α-globins and the β-globins, which are made in essentially equivalent amount in precursors of RBCs. Normal adult Hb (Hb A) has two α-globins and two β-globins (α2 β2). Genes on chromosomes 16 and 11 encode the α- and β-globins, respectively. There are also distinct embryonic, fetal, and minor adult analogs of the α- and β-globins, all of which are encoded by separate genes.  See Chapter 429 for a discussion of the developmental changes in Hb production.
See Chapter 429 for a discussion of the developmental changes in Hb production.
Disorders of Hb can be classified as qualitative or quantitative disorders. Qualitative abnormalities of Hb arise from mutations that change the amino acid sequence of the globin, thereby producing structural and functional changes in the Hb. There are four ways in which Hb can be qualitatively abnormal: (1) decreased solubility, (2) instability, (3) altered oxygen affinity, and (4) altered oxidation state of the heme-coordinated iron. Qualitative Hb disorders are often referred to as hemoglobinopathies, even though the term can technically apply to both qualitative and quantitative disorders. Quantitative Hb disorders result from the decreased and imbalanced production of generally structurally normal globins. For example, if β-globin production is diminished by a mutation, there will be a relative excess of α-globins. Such imbalanced production of α- and β-globins damages RBCs and their precursors in the bone marrow. These quantitative Hb disorders are called thalassemias. Both qualitative and quantitative disorders of Hb can be subdivided by the particular globin that is affected; for example, there can be α-thalassemias and β-hemoglobinopathies. We begin this chapter with a review of several of the common qualitative Hb disorders and end with a discussion of the thalassemias.
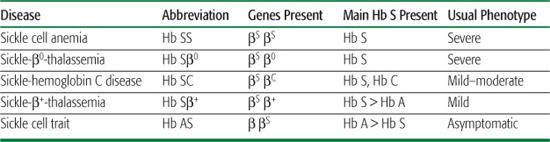
Table 434-1. Comparison of the Common Sickle Cell Diseases and Sickle Cell Trait
SICKLE CELL DISEASE
Sickle cell disease (SCD) is the name for a group of related disorders caused by sickle Hb (Hb S).1 Hb S is a qualitatively abnormal Hb caused by a point mutation of the β-globin gene.  This change decreases the solubility of Hb S in the deoxygenated state. Thus, as sickle red blood cells (RBCs) traverse the circulation, cycling through oxygenated and deoxygenated states, Hb S repeatedly forms rigid polymers that damage the RBC membrane, causing a hemolytic anemia and, ultimately, the manifestations of SCD.
This change decreases the solubility of Hb S in the deoxygenated state. Thus, as sickle red blood cells (RBCs) traverse the circulation, cycling through oxygenated and deoxygenated states, Hb S repeatedly forms rigid polymers that damage the RBC membrane, causing a hemolytic anemia and, ultimately, the manifestations of SCD.
 EPIDEMIOLOGY
EPIDEMIOLOGY
Sickle cell trait, the heterozygous or carrier state for the Hb S mutation, protects against severe malarial infections, so it arose and became a balanced polymorphism in historically malarious regions of the world. As such, sickle cell disease (SCD) occurs most commonly among individuals of African, Mediterranean, Middle Eastern, or Asian Indian ancestry. However, SCD is now found throughout the world in diverse populations, and it can occur in people of any skin color. In the United States, approximately 1 in 2500 newborns and 1 in 400 African American newborns has SCD. It is estimated that there are 100,000 individuals living with SCD in the United States.
 GENETICS AND PATHOPHYSIOLOGY
GENETICS AND PATHOPHYSIOLOGY
The prototypical, most common, and most severe form of sickle cell disease (SCD) is the homozygous state for the Hb S mutation (βS), called sickle cell anemia (Hb SS). Less common forms of SCD are compound heterozygous states that result from the coinheritance of the βS gene with other qualitatively or quantitatively abnormal β-globin genes. These include, in order of decreasing frequency, sickle-hemoglobin C disease (Hb SC), sickle-β+-thalassemia (Hb Sβ+), and sickle-β0-thalassemia (Hb Sβ0). Hb SS and Hb Sβ0 are clinically indistinguishable. Hb SC and Hb Sβ+ are, on average, milder forms of SCD.  Sickle cell trait, the carrier state, is not a form of SCD. See Table 434-1 to compare and contrast the different forms of SCD and sickle trait.
Sickle cell trait, the carrier state, is not a form of SCD. See Table 434-1 to compare and contrast the different forms of SCD and sickle trait.
As they traverse the circulation, red blood cells (RBCs) that contain mostly Hb S go through cycles of sickling (polymerization of Hb S) and unsickling (depolymerization of Hb S) due to deoxygenation of Hb S in the tissues and reoxygenation in the lungs. The tendency of individual RBCs to sickle is influenced by several factors. These include the presence of other Hbs that inhibit the polymerization of deoxy-Hb S, the hydration of the RBC, and the degree to which the Hb S is deoxygenated. When a solution of Hb S is deoxygenated, there is a characteristic delay time during which no polymerization occurs, followed by a phase of rapid polymerization. In vivo, this delay time allows most Hb S-containing RBCs to traverse the capillary beds before polymerization and sickling occur. A small number of RBCs remain permanently sickled due to membrane damage, even when fully oxygenated. These are called irreversibly sickled cells.
The two main pathophysiologic consequences of polymerization of Hb S, or sickling, are hemolysis and vasoocclusion (Fig. 434-1). Hemolysis, or destruction of RBCs, in SCD occurs predominantly in the extravascular compartment. Cycles of sickling damage the RBC, especially its membrane. These damaged RBCs are recognized as abnormal and removed from circulation by the reticuloendothelial system. Some intravascular hemolysis occurs as well, by microvascular trapping and destruction of adhesive and rigid sickle RBCs. The mean RBC life span in Hb SS is dramatically shortened to 10 to 20 days from the normal RBC life span of 120 days. The rate of hemolysis in SCD usually exceeds the rate at which new RBCs can be produced by the bone marrow. Therefore, SCD is characterized by a partially compensated hemolytic anemia.
In addition to their shortened life span, sickle erythrocytes are also abnormally adhesive and have decreased flexibility. Consequently, they can adhere to and damage the endothelium of blood vessels and block the flow of blood. This microvascular obstruction, called vasoocclusion, leads to ischemia and infarction of different tissues. Vasoocclusion is believed to be the main cause of acute episodes of pain, which are characteristic of SCD, as well as chronic organ damage.
 DIAGNOSTIC EVALUATION
DIAGNOSTIC EVALUATION
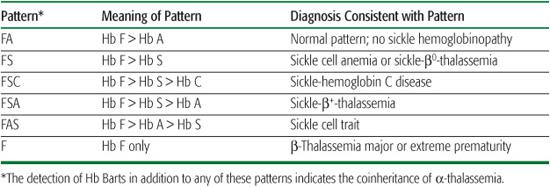
In the United States and some other countries, universal newborn screening programs for hemoglobinopathies now identify individuals with sickle cell disease (SCD) shortly after birth. Such early diagnosis is key to preventing early mortality from sepsis and acute splenic sequestration (see later in the chapter).  Results of newborn screening for hemoglobinopathies are usually reported as patterns, where all detected Hbs and their relative abundance are reported. All newborns, unless transfused, will have more Hb F than other Hbs. The remaining Hbs are reported in order of decreasing abundance. These patterns, in combination with confirmatory testing, permit the accurate, early diagnosis of SCD (Table 434-2).
Results of newborn screening for hemoglobinopathies are usually reported as patterns, where all detected Hbs and their relative abundance are reported. All newborns, unless transfused, will have more Hb F than other Hbs. The remaining Hbs are reported in order of decreasing abundance. These patterns, in combination with confirmatory testing, permit the accurate, early diagnosis of SCD (Table 434-2).
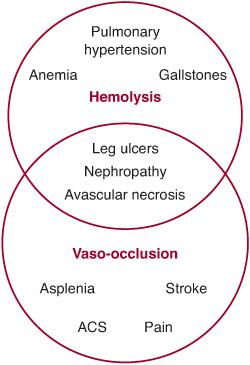
FIGURE 434-1. Overview of the pathophysiology of sickle cell disease (SCD) showing the hemolytic and vasoocclusive components and their overlap. ACS, acute chest syndrome.
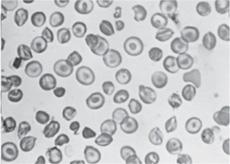
Beyond the immediate newborn period, the laboratory evaluation of suspected SCD should include a complete blood count, reticulocyte count, and examination of the peripheral blood smear (Fig. 434-2). Table 434-3 lists the usual hematologic findings in the common forms of SCD. To confirm a diagnosis of SCD, however, some analysis of Hb types must be performed. Abnormal Hbs must be identified using at least two methods because they can be difficult to differentiate.  Contemporary Hb separation methods include IEF and HPLC. DNA-based diagnostic methods, which are increasingly available, and family testing may be needed in occasional diagnostic challenges. It is important to know that a “sickle prep” or the Sickledex does not differentiate between SCD and sickle cell trait. These tests only confirm the presence of Hb S, which is found in both SCD and trait, so it is not helpful for the diagnosis of SCD.
Contemporary Hb separation methods include IEF and HPLC. DNA-based diagnostic methods, which are increasingly available, and family testing may be needed in occasional diagnostic challenges. It is important to know that a “sickle prep” or the Sickledex does not differentiate between SCD and sickle cell trait. These tests only confirm the presence of Hb S, which is found in both SCD and trait, so it is not helpful for the diagnosis of SCD.
Table 434-2. Common Patterns in Newborn Screening for Hemoglobinopathies
Because patients with SCD have a chronic hemolytic anemia (Table 434-3), they also have unconjugated hyperbilirubinemia and variable elevations of lactate dehydrogenase (LDH) and aspartate transaminase (AST).  It is generally not necessary to measure these substances. After the first year of life, the peripheral blood smear in Hb SS and Hb Sβ0 shows variable numbers of pathognomonic irreversibly sickled cells (Fig. 434-2), as well as polychromatophilic cells, Howell-Jolly bodies, poikilocytes, and target cells. Howell-Jolly bodies are nuclear remnants, and their presence on the blood smear outside the immediate neonatal period is indicative of hyposplenism. Patients with Hb SC and Hb Sβ+ usually do not have any irreversibly sickled cells, but they instead have a larger number of target cells. The mean cell volume (MCV) is normal in Hb SS and Hb SC, unless there is coinheritance of α-thalassemia. The MCV is low in Hb Sβ0 and Hb Sβ+. Leukocyte and platelet counts are usually moderately increased in SCD in the absence of infection.
It is generally not necessary to measure these substances. After the first year of life, the peripheral blood smear in Hb SS and Hb Sβ0 shows variable numbers of pathognomonic irreversibly sickled cells (Fig. 434-2), as well as polychromatophilic cells, Howell-Jolly bodies, poikilocytes, and target cells. Howell-Jolly bodies are nuclear remnants, and their presence on the blood smear outside the immediate neonatal period is indicative of hyposplenism. Patients with Hb SC and Hb Sβ+ usually do not have any irreversibly sickled cells, but they instead have a larger number of target cells. The mean cell volume (MCV) is normal in Hb SS and Hb SC, unless there is coinheritance of α-thalassemia. The MCV is low in Hb Sβ0 and Hb Sβ+. Leukocyte and platelet counts are usually moderately increased in SCD in the absence of infection.
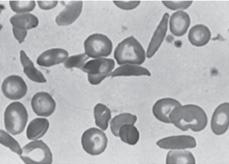
FIGURE 434-2. Peripheral blood film in sickle cell anemia (Hb SS).
 CLINICAL COURSE
CLINICAL COURSE 
A patient with SCD has at baseline a chronic hemolytic anemia to which he or she becomes physiologically adapted. This baseline anemic but relatively healthy state is called the steady state. This steady state is punctuated by intermittent, acute episodes of illness, called vasoocclusive episodes, events, or crises. Recurrent vasoocclusion and chronic anemia also produce chronic organ damage. There are a number of different types of acute episodes and chronic organ damage that are described in this chapter. Patients with any form of SCD can experience these complications, but they tend to occur earlier and more frequently in Hb SS and Hb Sβ0.
At birth, newborns with Hb SS disease have normal birth weight and are not anemic. Anemia and reticulocytosis usually appear between 2 and 6 months of age. Along with the anemia come jaundice and a cardiac flow murmur. Jaundice and flow murmurs are expected findings that should not cause concern. Splenic infarction and hyposplenism may begin to occur by 3 months of age. Therefore, it is necessary to prescribe prophylactic penicillin before this time to prevent pneumococcal sepsis. Before splenic infarction is complete, the spleen may be palpably enlarged. Nevertheless, it is still poorly or nonfunctional. Acute vasoocclusive events are unusual before 6 months of age. The first painful event is often dactylitis, which is a painful swelling of the hands and feet. Dactylitis is rare beyond 3 years of age. Although birth weight is normal, growth retardation is commonly observed during childhood. On average, individuals with Hb SS tend to be thinner and shorter than their peers, but a normal adult height can often be attained. The onset of puberty and development of secondary sex characteristics are usually delayed by 2 to 3 years.
The course of the disease does not improve with age, but certain complications occur more commonly in adolescents and adults, such as retinal disease, leg ulcers, and pulmonary hypertension. The median life expectancy for patients with Hb SS and Hb Sβ0 in developed nations is now estimated to be 53 years for men and 58.5 years for women. Patients with Hb SC and Hb Sβ+ have a near normal life expectancy. In many Third World countries, 90% of children with Hb SS disease do not survive beyond 5 years of age, and falciparum malaria is the leading cause of death. Although sickle cell trait offers protection against malaria, the combination of Hb SS and malaria is usually fatal.
The common acute complications of sickle cell disease in childhood and some of the forms of chronic organ damage and dysfunction that occur with time are presented.
Acute Episodes: Infections
Until the end of the 20th century, infections were the major cause of death in young children with Hb SS and Hb Sβ0 in the United States. Fatal Streptococcus pneumoniae sepsis occurred in 15% to 20% of children in the first 5 years of life. These infections were typically fulminant, with death occurring within 24 hours of the onset of fever. Children with sickle cell disease (SCD) have an unusual vulnerability to severe pneumococcal sepsis due to their early loss of splenic reticuloendothelial function (functional hyposplenism) and their lack of circulating antibodies against polysaccharide-encapsulated bacteria. In early life, the spleen, although often palpably enlarged, loses reticuloendothelial function because of continuous vasoocclusive infarction. The enlarged spleen gradually becomes small and fibrotic, and it is rarely palpable after 6 years of age. As a normal developmental phenomenon, young children cannot efficiently produce antibodies against polysaccharide antigens, such as those on the capsule of the pneumococcus. When circulating antipneumococcal antibodies are lacking, the spleen is almost exclusively responsible for the clearance of pneumococci in the circulation. The combination of hyposplenism and lack of antibodies against polysaccharide antigens accounts for the high susceptibility and the historically high frequency and mortality of pneumococcal infections in young children with Hb SS and Hb Sβ0.
Table 434-3. Typical Laboratory Findings in the Common Forms of Sickle Cell Disease

Fatal pneumococcal sepsis is now rare in children with SCD because of universal newborn screening for hemoglobinopathies, prophylactic penicillin, and use of the conjugated pneumococcal vaccine. Newborn screening programs identify children with SCD before they develop hyposplenism. This allows time for physicians to teach parents to seek medical attention for any high fever (generally 101.5°F or more) and to initiate prophylactic penicillin. A national placebo-controlled study (PROPS) showed that oral penicillin prophylaxis reduced the incidence of invasive pneumococcal infections by 84% in Hb SS children younger than 5 years. A follow-up study (PROPS-II) did not show effectiveness of continuing penicillin prophylaxis after 5 years of age, so most centers discontinue prophylaxis at this age. Compared to Hb SS and Sβ0, individuals with Hb SC and Hb Sβ+ have a much lower risk of pneumococcal sepsis because their splenic infarction is delayed and incomplete, so prophylactic penicillin is generally not necessary for these patients. The newer heptavalent conjugated pneumococcal vaccine (Prevnar) has further decreased the incidence of pneumococcal infection, and it is recommended for all children with SCD and given according to guidelines for all children. The older, 23-valent polysaccharide vaccine (eg, Pneumovax) should be given at age 2 and 5 years. Although meningococcal infections have been reported only rarely in SCD, it might also be prudent to administer the newer conjugated meningococcal vaccine (Menactra) to patients with SCD.
Patients with SCD also have a predilection for osteomyelitis  . Salmonella species cause about half the cases of osteomyelitis in SCD and staphylococci most of the rest. It can be difficult to differentiate osteomyelitis from an acute painful episode that results in infarction of cortical bone. Both can cause bony tenderness, effusions, and lucencies on roentgenograms. Even bone scans and magnetic resonance imaging fail to distinguish between the two. Clinical features are more helpful than imaging studies in this situation. The occurrence of fever, a single focus of pain, and a positive blood culture are more consistent with a diagnosis of osteomyelitis than an acute painful episode.
. Salmonella species cause about half the cases of osteomyelitis in SCD and staphylococci most of the rest. It can be difficult to differentiate osteomyelitis from an acute painful episode that results in infarction of cortical bone. Both can cause bony tenderness, effusions, and lucencies on roentgenograms. Even bone scans and magnetic resonance imaging fail to distinguish between the two. Clinical features are more helpful than imaging studies in this situation. The occurrence of fever, a single focus of pain, and a positive blood culture are more consistent with a diagnosis of osteomyelitis than an acute painful episode.
Acute Episodes: Splenic Sequestration
In young children in whom splenic autoinfarction is not yet complete, the spleen may become acutely enlarged and engorged with blood sequestered from the systemic circulation, with consequent severe anemia, hypovolemia, and marked splenic enlargement. Thrombocytopenia usually occurs, as well, due to hypersplenism. The recognition of acute splenic enlargement and the signs and symptoms of acutely severe anemia by both parents and health care professionals is important to prevent a fatal outcome. Parents can be taught how to palpate the spleen size and to seek medical attention for any sudden enlargement. Rapid transfusion of blood is the most important intervention for severe anemia and hypovolemic shock due to acute splenic sequestration. Not all sequestration is severe. Some episodes are characterized by only mild decreases in hemoglobin concentration (1–2 g/dL) and modest increases in spleen size. These may be associated with viral infections and require only careful observation. Splenic sequestration is often recurrent, and splenectomy is often advocated after recovery from a severe episode or recurrent episodes of lesser severity. Because the spleen is usually not functional, splenectomy does not appear to further increase the risk of infection.
Acute Episodes: Aplastic Crisis
Red blood cell (RBC) life span is greatly shortened from the normal of 120 days to 10 to 20 days in Hb SS and Hb Sβ0. Consequently, patients must chronically maintain a marked increase in RBC production by the bone marrow, producing a chronic reticulocytosis, to maintain a stable Hb concentration compatible with life. If RBC production is impaired, for even a short time, the Hb concentration will fall rapidly. During many viral infections and inflammatory states, erythropoiesis may be modestly reduced, resulting in relative reticulocytopenia and transiently more severe anemia. Human parvovirus, the agent of fifth disease, has especially profound effects on patients with chronic hemolytic anemia. Parvovirus destroys early RBC precursors in the bone marrow and causes RBC aplasia for about a week. The consequent erythroblastopenia and reticulocytopenia cause a dramatic and potentially life-threatening anemia, with the Hb concentration falling as low as 1 to 2 g/dL without measurable reticulocytes. This episode of severe anemia is called the aplastic crisis. Provided the patient does not die, the acute RBC aplasia is transient and self-limited. Spontaneous recovery begins about 1 week after the onset of reticulocytopenia due to antibody-mediated clearance of the virus. Recovery is heralded by the appearance of nucleated RBCs in the circulation, followed shortly by a brisk reticulocytosis. Transfusion of blood is the most important intervention for symptomatic or severe anemia. In contrast to acute splenic sequestration, blood must be transfused relatively slowly to patients with aplastic crisis because their blood volume is typically normal or increased as a physiologic adaption to a more slowly progressive anemia. Lifelong immunity against parvovirus prevents recurrent episodes. Patients with Hb SC and Sβ+ can also have aplastic crisis, but their anemia tends to be less severe because their RBC life span is longer than Hb SS and Sβ0 patients. 
Acute Episodes: Painful Episodes
The acute painful episode is the hallmark of sickle cell disease (SCD). It is the most common reason for medical consultation and hospitalization in this population. The pain seems to be caused by acute vasoocclusion, primarily in bones and bone marrow, with consequent ischemia and inflammation. Pain can occur anywhere in the body, but it is most commonly osteoarticular and juxtavertebral. The earliest physical manifestation of Hb SS is often a characteristic painful episode called dactylitis. Dactylitis is an often symmetric swelling of the hands and feet that occurs in about 30% of children in the first 3 years of life. As the child ages, painful episodes involve instead the long bones, vertebrae, sternum, ribs, lower back, and abdomen. Unlike dactylitis, most other painful episodes typically do not have accompanying physical signs. Infection, dehydration, and exposure to the cold may precipitate pain, but specific precipitating features are usually not identified. Although painful events severe enough to require hospitalization are infrequent (< 1/year) for most patients, less severe painful events that can be managed at home with oral analgesics occur more frequently. 
The treatment of the painful episode is symptomatic—controlling the pain. Analgesia must be tailored to the degree of pain and the patient. Moderate pain without fever or other signs of concomitant illness can usually be managed at home with hydration, analgesics, and rest. However, some pain is so severe that hospitalization for intravenous hydration and parenteral opioids is necessary. Overhydration is not helpful, and it may actually precipitate acute chest syndrome. A combination of nonsteroidal anti-inflammatory drugs and opioid analgesics, titrated to effect, will usually achieve adequate pain relief. Patients with SCD-related pain, even severe, typically have no accompanying physical signs, such as edema or erythema, so it is important to believe the patient’s report of pain and its intensity. Transfusion does not relieve acute sickle cell pain, and there is no therapy to shorten the duration of the painful episode. The pain must be treated until the episode resolves spontaneously, which may take as long as a week. Once the pain begins to resolve, opioid therapy can be decreased relatively quickly.
Transfusion does not relieve acute sickle cell pain, and there is no therapy to shorten the duration of the painful episode. The pain must be treated until the episode resolves spontaneously, which may take as long as a week. Once the pain begins to resolve, opioid therapy can be decreased relatively quickly.
Acute Episodes: Acute Chest Syndrome
Acute chest syndrome is a descriptive term for almost any acute pulmonary illness in a child with sickle cell disease (SCD). It is defined practically as a new pulmonary infiltrate with some combination of fever, cough, chest pain, tachypnea, dyspnea or hypoxemia. Acute chest syndrome has many inciting causes, including pulmonary infection, infarction, atelectasis, overhydration causing pulmonary edema, bronchospasm and airway inflammation, or fat embolism from bone marrow infarction. Acute chest syndrome often starts as a small infiltrate in one lobe, but it can progress rapidly to involve multiple lobes, resulting in respiratory distress severe enough to require intubation and ventilatory support. Acute chest syndrome is a leading cause of death in adolescents and adults. Management includes oxygen supplementation for hypoxemia, maintenance of hydration without overhydration, adequate but not excessive analgesia, and antibacterials. An infectious etiology is often not apparent; however, the use of empiric antibiotics active against pneumococcus, Mycoplasma, and Chlamydia is prudent. Simple RBC transfusions or exchange transfusions may be needed for moderate and especially severe cases of acute chest syndrome. 
Acute Episodes: Stroke
Clinically overt stroke occurs in about 11% of patients with Hb SS by 18 years of age if they do not receive primary stroke prophylaxis.  The reason for the high incidence in young children is unknown. There are three main clinical presentations of overt cerebrovascular disease in Hb SS: cerebral infarction, cerebral hemorrhage, and transient ischemic attacks (TIAs). Infarction and hemorrhage cause weakness, paralysis, and aphasia, and sometimes seizures and headache. TIAs are episodes of weakness, paralysis, or aphasia that last less than 24 hours. TIAs are likely to be overlooked in young children, but they are harbingers of overt stroke. All three presentations can occur at any age, but hemorrhagic stroke is more common in patients older than 20 years. In young children, stenosis of the large intracerebral blood vessels is the usual antecedent of stroke. Magnetic resonance imaging (MRI) and magnetic resonance angiography (MRA) are needed to visualize cerebral infarction and stenosis of intracranial arteries. Computerized tomography (CT) can be obtained more quickly than MRI, and it may be helpful when cerebral hemorrhage is strongly suspected. Acute management includes exchange transfusion, careful hydration, and control of any seizures. An additional 20% to 30% of patients experience covert or “silent” cerebral infarctions that are not accompanied by motor signs. Both overt and covert strokes can cause neurocognitive impairment.
The reason for the high incidence in young children is unknown. There are three main clinical presentations of overt cerebrovascular disease in Hb SS: cerebral infarction, cerebral hemorrhage, and transient ischemic attacks (TIAs). Infarction and hemorrhage cause weakness, paralysis, and aphasia, and sometimes seizures and headache. TIAs are episodes of weakness, paralysis, or aphasia that last less than 24 hours. TIAs are likely to be overlooked in young children, but they are harbingers of overt stroke. All three presentations can occur at any age, but hemorrhagic stroke is more common in patients older than 20 years. In young children, stenosis of the large intracerebral blood vessels is the usual antecedent of stroke. Magnetic resonance imaging (MRI) and magnetic resonance angiography (MRA) are needed to visualize cerebral infarction and stenosis of intracranial arteries. Computerized tomography (CT) can be obtained more quickly than MRI, and it may be helpful when cerebral hemorrhage is strongly suspected. Acute management includes exchange transfusion, careful hydration, and control of any seizures. An additional 20% to 30% of patients experience covert or “silent” cerebral infarctions that are not accompanied by motor signs. Both overt and covert strokes can cause neurocognitive impairment.
Overt stroke has a 50% to 90% risk of recurrence, so secondary stroke prophylaxis is used to prevent further brain injury. Recurrence can be lowered to approximately 10% to 20% by chronic RBC transfusions. Chronic transfusions usually aim to maintain the percentage of Hb S in the blood at less than 30%. Although chronic transfusions can effectively prevent recurrent stroke in most children, complications include iron overload and the need for chelation therapy, alloimmunization, and transfusion-transmitted infections. An increasingly popular therapeutic strategy is to prevent first strokes. Transcranial Doppler (TCD) ultra-sonography is a noninvasive screening tool that can detect stenosis in the major cerebral arteries and thereby identify the children at highest risk of overt stroke. Children with abnormal TCD measurements can be offered chronic transfusions for primary stroke prophylaxis. It is not yet known when or if chronic transfusions can ever be stopped in primary or secondary stroke prophylaxis. It is also not yet known whether covert strokes can be prevented by chronic transfusions. These are areas of active clinical investigation.
Acute Episodes: Priapism
Priapism is a painful, prolonged erection of the penis. It may occur starting in early childhood, and the estimated prevalence in males with Hb SS is 30% to 40% by 18 years of age. Episodes occur in two patterns: prolonged, lasting more than 4 hours, and shorter “stuttering” episodes. Stuttering priapism may occur in clusters and sometimes daily. A prolonged episode of priapism requires rapid intervention to prevent ischemic damage to the penis and erectile dysfunction. Management includes hydration, pain control, and oral adrenergic medications such as pseudoephedrine. If an episode lasts longer than 4 hours, aspiration of the corpora cavernosa and irrigation with a dilute solution of phenylephrine or epinephrine is indicated. Chronic transfusion may prevent recurrent events, but the use of simple transfusion for an acute event is unproven.
Chronic Organ Dysfunction and Damage
In addition to early splenic dysfunction and involution, other forms of progressive organ dysfunction or damage occur with increasing age in the kidneys, bones, eyes, lungs, heart, and liver. Renal complications begin early in life with hyposthenuria, a defect in renal concentration. The consequent production of dilute urine causes enuresis and predisposes to dehydration. Other renal complications include papillary necrosis and hematuria, proteinuria, and renal failure. Renal failure is uncommon in children. Avascular necrosis of bone from vasoocclusion, especially of the heads of the femur and humerus, may develop in some patients and causes chronic bone pain. Occlusion of retinal vessels may lead to proliferative retinopathy, retinal detachment, and loss of vision in adolescents and adults. Chronic hemolytic anemia and, perhaps, pulmonary infarction may cause pulmonary hypertension, hypoxemia, and right-sided heart failure. Pulmonary hypertension is a risk factor for early death in adults with sickle cell disease (SCD). High-output, left-sided heart failure may occur in patients with more severe degrees of anemia. Cholelithiasis secondary to chronic hemolysis can occur as early as 3 to 4 years of age, and more than half of adults with Hb SS have gallstones. Cholecystectomy is one of the most common surgical procedures performed in patients with SCD. Hepatomegaly and hepatic dysfunction or failure may occur as a result of sequestration, infarction, hepatobiliary stasis, or infection.
 TREATMENT
TREATMENT
There are three main disease-modifying treatments that can reduce the overall severity of sickle cell disease (SCD) or cure it: hydroxyurea, chronic transfusions, and hematopoietic stem cell transplantation. Hydroxyurea is a cytostatic chemotherapeutic agent that has multiple beneficial effects in patients with SCD. Hydroxyurea increases the concentration of Hb F, decreases the leukocyte count, decreases the platelet count, and improves blood rheology. Clinically, hydroxyurea reduces the frequency of painful crises, acute chest syndrome, and transfusions by about 50% in adults. Smaller studies in children have shown similar effects. The long-term safety of hydroxyurea in patients with SCD of any age, and especially those younger than 5 years, has not been clearly established.
Chronic transfusion programs entail regular, usually monthly, transfusions of packed RBCs with the aim to maintain the percentage of Hb S in the blood less than 30%. Chronic transfusions are effective at preventing most complications of SCD, but the most common indications are primary and secondary stroke prophylaxis. Complications of transfusions include iron overload and the need for chelation therapy, alloimmunization, and transfusion-transmitted infections.
Stem cell (or bone marrow) transplantation is the only cure for SCD. Widespread use of transplantation is limited by the lack of donor availability and toxicities of the procedure. Transplantation is safest when hematopoietic stem cells are obtained from an human leucocyte antigen (HLA)-matched sibling (without SCD), but only 10% of patients actually have a potential donor. Transplantation is associated with mortality rate of about 5%, a graft rejection rate of about 5%, and the occurrence of significant graft-versus-host disease in about 5%. Therefore, the chances of a successful, curative transplant without major morbidity are around 85% to 90%. In North America, the most common indication has been stroke or cerebrovascular disease, but transplantation is also offered for other, especially recurrent, vasoocclusive complications. 
 SICKLE CELL TRAIT
SICKLE CELL TRAIT
Sickle cell (Hb S) trait is the carrier or heterozygous state for the βS gene; the other allele encodes a normal β-globin. Hb S trait is very common: 1 in 12 African Americans has it. Hb S trait is not a form of sickle cell disease (SCD); carriers have normal life expectancy, blood counts, RBC indices, reticulocyte counts, and peripheral smears. Hb electrophoresis shows more Hb A (60–70%) than Hb S (30–40%) in Hb S trait, in contrast to Hb Sβ+ where there is more Hb S than Hb A (Tables 434-1 and 434-2). The predominance of Hb A within Hb S trait RBCs prevents sickling under normal oxygen tensions. The hypertonicity and relative acidosis in the renal medulla can induce sickling in the kidney, however. Therefore, hyposthenuria and renal papillary necrosis with gross hematuria are known potential medical complications of Hb S trait. 
It is important for individuals to know that they have Hb S trait because of the risk to their offspring. Hgb SS is an autosomal-recessive disease, so if both parents have Hb S trait, then each of their offspring will have a 25% chance of having Hb SS. Even if only one parent has Hb S trait, offspring are still at risk of having SCD if the parent without Hb S trait happens to have Hb C trait or β-thalassemia trait, for example. Nondirective genetic counseling should be offered to adolescents and adults with Hb S trait. Preimplantation and prenatal diagnosis are available.
HEMOGLOBIN C
Like Hb S, Hb C is a qualitative abnormality of the β-globin. Hb C originated in West Africa, and Hb C trait is found in about 2.5% of African Americans. Individuals with Hb C trait have no hematologic abnormalities, except for increased numbers of target cells on the peripheral blood smear and the presence of 30% to 40% the slowly migrating Hb C on electrophoresis. Homozygotes for Hb C (Hb CC disease) have a mild hemolytic anemia and splenomegaly, but do not have vasoocclusive symptoms, splenic infarction, or hyposplenism. Hb CC disease is not a form of sickle cell disease. The red blood cells are microcytic, and the blood smear shows many target cells, folded cells, and Hb C crystals (Fig. 434-3). The compound heterozygous state, Hb C-β-thalassemia, is similar to Hb CC disease.
Hb C originated in West Africa, and Hb C trait is found in about 2.5% of African Americans. Individuals with Hb C trait have no hematologic abnormalities, except for increased numbers of target cells on the peripheral blood smear and the presence of 30% to 40% the slowly migrating Hb C on electrophoresis. Homozygotes for Hb C (Hb CC disease) have a mild hemolytic anemia and splenomegaly, but do not have vasoocclusive symptoms, splenic infarction, or hyposplenism. Hb CC disease is not a form of sickle cell disease. The red blood cells are microcytic, and the blood smear shows many target cells, folded cells, and Hb C crystals (Fig. 434-3). The compound heterozygous state, Hb C-β-thalassemia, is similar to Hb CC disease.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


