Previous
Proposed
Intersex
DSD
Male pseudohermaphrodite, undervirilization of a Y male, and undermasculinization of an XY male
46, XY DSD
Female pseudohermaphrodite, overvirilization of an XX female, and masculinization of an XX female
46, XX DSD
True hermaphrodite
Ovotesticular DSD
XX male or XX sex reversal
46, XX testicular DSD
XY sex reversal
46, XY complete gonadal dysgenesis
Embryology
Normal sexual differentiation and subsequent development depend on the genetic sex (XX or XY) .
Until 7 weeks of gestation, the fetus has undetermined sex with indifferent gonads (bipotential gonad) and internally developing both Wolffian and Müllerian ducts.
Various genes expressed by the Y chromosome are responsible for the differentiation of the bipotential gonad to develop into testes.
A 35-kilobase pair (kbp) gene (testis-determining factor, TDF) on the 11.3 subband of the distal short arm of the Y chromosome, an area termed as the sex-determining region of the Y chromosome (SRY), is responsible for initiating differentiation of the bipotential gonad and testes formation.
The SRY also codes for a transcription factor that acts in the somatic cells of the genital ridge. This triggers a cascade of events that leads to the development of testicular Sertoli and Leydig cells.
The SRY expression directs testicular morphogenesis, leading to the production of Müllerian-inhibiting substance (MIS) by the Sertoli cells and, later, testosterone by the Leydig cells.
Surprisingly, more than half of the patients with XX ovotesticular DSD lack SRY, despite the presence of testicular differentiation. This suggests that this gene codes for a product that reacts with other genes on Y, X, and/or autosomes to complete testicular differentiation .
In 46, XY males, the Sertoli cells of the testes are responsible for the production of MIS, which causes regression of the Müllerian ducts. The Leydig cells then produce testosterone which induces the primordial Wolffian (mesonephric) duct to develop into the epididymis, vas deferens, and seminal vesicle.
MIS is produced by the Sertoli cells of the testis. It is a protein with a molecular weight of 15,000 d that is secreted by the testis beginning in the eighth fetal week. In a male fetus with normal testicular function, MIS represses Müllerian duct development (Fallopian tubes, uterus, and upper vagina) .
In the fetus with 46, XX, female gonadal development ensues.
Ovarian differentiation from the bipotential gonad appears to rely on a mechanism that is triggered mostly, but not solely, by the absence of the SRY.
Internal genitalia of the female fetus develop if there is no exposure to the SRY gene and its signaling molecules. The Wolffian duct (because of absence of SRY) regresses and the Müllerian duct (because of absence of MIS) then matures into the oviduct, uterus, cervix, and upper vagina.
Hormone expression during the ninth week of gestation, from the testes or ovary, stimulates external genitalia development. By the 14th week of gestation, the external genitalia have been formed.
The external genitalia of both sexes are identical during the first 7 weeks of gestation. Without the hormonal action of the androgens, testosterone, and dihydrotestosterone (DHT), external genitalia appear phenotypically female (Fig. 66.1)
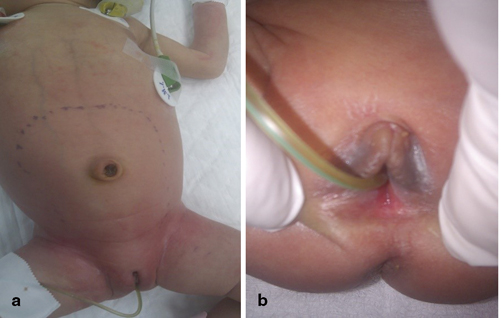
Fig. 66.1
a and b Clinical photographs of a newborn with vaginal atresia causing hydrocolpos
In the gonadal male:
Differentiation toward the male phenotype occurs over the next 8 weeks .
This is under the influence of testosterone, which is converted to 5-DHT by the action of 5-alpha reductase enzyme, present within the cytoplasm of cells of the external genitalia and the urogenital sinus.
DHT is bound to cytosol androgen receptors within the cytoplasm and is subsequently transported to the nucleus, where it leads to translation and transcription of genetic material.
This in turn leads to normal male external genital development from primordial parts, forming the scrotum from the genital swellings, forming the shaft of the penis from the folds, and forming the glans penis from the tubercle.
The prostate develops from the urogenital sinus.
The timing of this testosterone-related developmental change begins at approximately 6 weeks of gestation, with a testosterone rise in response to a surge of luteinizing hormone (LH).
Testosterone levels remain elevated until the 14th week.
Most phenotypic differentiation occurs during this period .
After the 14th week, fetal testosterone levels settle at a lower level, and are maintained more by maternal stimulation through human chorionic gonadotropin (HCG) than by LH .
Testosterone’s continued action during the latter phases of gestation is responsible for continued growth of the phallus, which is directly responsive to testosterone and to DHT (Fig. 66.2).
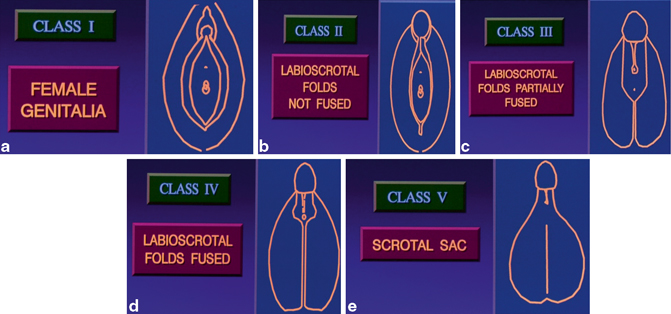
Fig. 66.2
a–e Diagrammatic representation of embryological differentiation of the external genitalia between males and females
Incomplete masculinization occurs:
When testosterone fails to convert to DHT
When DHT fails to act within the cytoplasm or nucleus of the cells of the external genitalia and urogenital sinus
During the developmental process, there are multiple opportunities for errors in differentiation, all of which are possible causes of the DSD.
In humans, genetic sex has traditionally been evaluated through establishing the karyotype of peripheral lymphocytes.
The peripheral karyotypes of patients with ovotestes-DSD show marked variation:
◦ Approximately 60 % are 46, XX.
◦ 15 % are 46, XY.
◦ 25 % show various forms of mosaicism .
◦ Less than 1 % show 46, XX/46, XY chimerism or the existence of two or more cell lines, each of which has a different genetic origin.
The existence of patients with 46, XX DSD, who have testicular tissue in the absence of a Y chromosome or SRY, clearly shows that other genetic components are involved. Other genes have been shown to play a role in testicular development. These include DAX1 on the X chromosome, S.1 on band 9q33, WT1 on band 11p13, SOX9 on bands 17q24-q25, and AMH on band 19q13.3. Fetal ovaries develop when the TDF gene (or genes) is absent .
Classification
DSD classification is based on the new nomenclature as shown in Tab. 66.2 .
Tab. 66.2
DSD classifications
Sex chromosome DSD | 46, XY DSD | 46, XX DSD |
|---|---|---|
45, X (Turner syndrome and variants) | Disorders of testicular development: 1. Complete gonadal dysgenesis (Swyer syndrome) 2. Partial gonadal dysgenesis 3. Gonadal regression 4. Ovotesticular DSD | Disorders of gonadal (ovarian) development: 1. Ovotesticular DSD 2. Testicular DSD (e.g., SRY+, duplicate SOX9) 3. Gonadal dysgenesis |
47, XXY (Klinefelter syndrome and variants) | Disorders in androgen synthesis or action: 1. Androgen biosynthesis defect (e.g., 17-hydroxysteroid dehydrogenase deficiency, 5αRD2 deficiency, StAR mutations) 2. Defect in androgen action (e.g., CAIS, PAIS) 3. Luteinizing hormone receptor defects (e.g., Leydig cell hypoplasia, aplasia) 4. Disorders of anti-Müllerian hormone (AMH) and AMH receptor (persistent Müllerian duct syndrome) | Androgen excess: 1. Fetal (e.g., 21-hydroxylase deficiency, 11-hydroxylase deficiency) 2. Fetoplacental (aromatase deficiency, P450 oxidoreductase [POR]) 3. Maternal (luteoma, exogenous, etc.) |
45, X/46, XY (MGD, ovotesticular DSD) | Other (e.g., cloacal exstrophy, vaginal atresia, Müllerian, renal, cervicothoracic somite abnormalities [MURCS], other syndromes) | |
46, XX/46, XY (ovotesticular DSD) |
CAIS complete androgen insensitivity syndrome, PAIS partial androgen insensitivity syndrome
Sex chromosome DSD :
45, X (Turner syndrome and variants)
47, XXY (Klinefelter syndrome and variants)
45, X/46, XY (MGD, ovotesticular DSD)
46, XX/46, XY (chimeric, ovotesticular DSD)
46, XY DSD: chromosomally male, but DSD results from :
Disorders of testicular development (complete and partial gonadal dysgenesis)
Disorders of androgen synthesis or action (complete and partial androgen insensitivity, disorders of AMH/anti-Müllerian receptor, androgen biosynthesis defect)
Syndromes associated with defects in genital development, including cloacal anomalies, vanishing testes syndrome, congenital hypogonadotropic hypogonadism, and severe hypospadias
46, XX DSD: chromosomally female, but DSD results from :
Disorders of ovarian development (ovotesticular DSD, testicular DSD, gonadal dysgenesis)
Disorders leading to androgen excess (fetal, e.g., CAH, fetoplacental, maternal)
Abnormalities of Müllerian ducts (Müllerian ducts dysgenesis or hypoplasia, vaginal atresia, cloacal exstrophy; Fig. 66.3) .
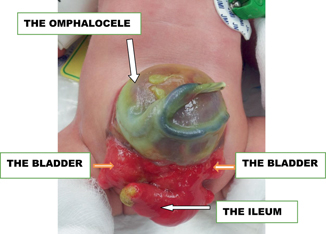
Fig. 66.3
A clinical photograph of a patient with cloacal extrophy showing distorted anatomy of external genitalia
Etiology
The etiology of DSD that is present at birth depends on the type of disorder (Fig. 66.4) .
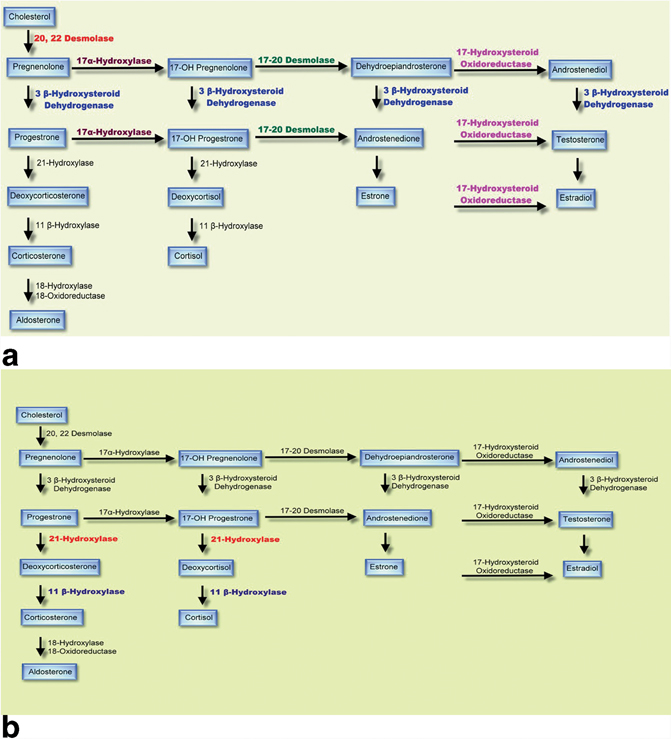
Fig. 66.4
a Diagrammatic representation of steroid biosynthesis. b The enzymes likely to be affected in congenital adrenal hyperplasia
Most causes of DSD are genetic.
One exception is virilization of a 46, XX fetus due to maternal virilizing tumors or maternal ingestion of androgenic drugs .
Genetic causes include:
1.
Sex chromosome DSD:
◦ This presents as ambiguous genitalia due to meiotic or mitotic gain or loss of a chromosome, or due to chimerism.
◦ 45X/46, XY MGD is due to postmeiotic errors, leading to loss of a Y chromosome in one cell lineage.
◦ 46, XX/46, XY chimerism is due to the fusion of two zygotes.
2.
46, XY DSD:
◦ Gonadal dysgenesis may be due to mutations in one of several genes that are involved in testicular development.
◦ Defects in SRY can cause ovotesticular DSD.
◦ Disorders of androgen synthesis which include mutations in genes responsible for testosterone synthesis. These can be common to the adrenal gland and the testes, and thus cause deficiencies of glucocorticoids and/or mineralocorticoids or are just found in the testes and thus only lead to defects in testosterone biosynthesis. These include :
◦ 17-hydroxylase deficiency and 20–22 lyase deficiency due to mutations in CYP17.
◦ 3-hydroxysteroid dehydrogenase deficiency due to mutations in HSD3B2.
◦ 5-alpha-reductase deficiency due to mutations in steroid 5-alpha-reductase (SRD5A2), which converts testosterone to DHT.
◦ Steroidogenic acute regulatory (StAR) protein deficiency.
◦ These are all autosomal-recessive conditions.
◦ Partial androgen insensitivity syndrome is due to mutations in the androgen receptor on Xq11. This is an X-linked recessive condition.
◦ Various genetic syndromes associated with defects in genital development, such as Smith–Lemli–Opitz syndrome
◦ Congenital hypogonadotropic hypogonadism can be due to one of several gene defects .
3.
46, XX DSD:
◦ CAH.
◦ CAH is due to 21-hydroxylase deficiency (CYP21A2 on chromosome 6p21.3).
◦ This is the cause in more than 95 % of cases.
◦ This is an autosomal-recessive condition.
◦ Some of the other causes are 11-beta-hydroxylase deficiency (CYP11B1 on chromosome 8q21).
◦ 3-beta-hydroxysteroid dehydrogenase deficiency (HSD3B2 on chromosome1p13.1).
◦ Gonadal dysgenesis can be due to translocation of an SRY gene onto an X chromosome or to duplication of the SOX9 region .
Pathophysiology
MGD
Characterized by asymmetrical development of the gonads and internal genital ducts .
There is a dysgenetic testis and thus poorly developed Wolffian ducts on one side and a streak gonad (ovary) on the other side.
Variable degrees of external genital ambiguity are found.
The lack of a Y chromosome, and thus lack of the SRY gene, on one side presumably leads to the dysgenetic testis that produces some, but not enough, testosterone for normal male development.
The degree of virilization depends on the degree of functioning testes.
In 46, XY DSD presenting as ambiguous genitalia, the degree of virilization depends on:
The amount of residual functioning testes.
The amount of testosterone synthesized.
The degree of androgen insensitivity.
In 46, XX DSD with CAH due to 21-hydroxylase deficiency :
The degree of virilization depends on the residual activity of the 21-hydroxylase gene.
In the absence of any 21-hydroxylase activity, essentially all of the cortisol precursors are directed to androgen production leading to high androgen levels and severe virilization. These cases are associated with salt wasting.
Evaluation of a Newborn with DSD
The diagnostic approach to a newborn with ambiguous genitalia involves a multidisciplinary team approach .
This includes a pediatric endocrinologist, geneticist, pediatric surgeon, pediatric urologist, and a neonatologist.
Sex assignment.
A sex is not assigned to the baby until the evaluation has been completed, but as soon as is feasible.
The child is referred to as “baby,” not boy or girl.
The family should be encouraged to delay naming the baby until the sex has been assigned .
The needs, background, culture, and expectations of the parents must be understood and respected.
Parents should understand that children with a DSD can live normal lives and function well in society.
Educating the parents regarding sexual development is also important, as this helps them understand and participate in the process.
The most common disorder of DSD, CAH, results in virilization of a 46, XX female and thus is classified under the group of 46, XX DSD. It is important to distinguish CAH from other less common causes of DSD.
Although different disorders may present with similar findings on physical examination, there are often aspects of the examination that are crucial and will help define the disorder and may help direct the necessary investigation .
It is important to rule out a malformation syndrome in a patient with ambiguous genitalia.
Gonadal and external genitalia examination:
Note the size and degree of differentiation of the phallus, since variations may represent clitoromegaly or hypospadias.
Phallus length:
◦ A normal-term male penis is 3.5 ± 0.7 cm.
◦ A length of less than 2.0 cm is considered abnormal .
◦ A normal-term female clitoris is less than 1.0 cm.
◦ Micropenis is thus defined as a stretch penile length of less than 2.0 cm in a term male infant, and clitoromegaly as a clitoris greater than 1.0 cm in a term female. In preterm infant males, the penile length is shorter.
Note the position of the urethral meatus (Fig. 66.5).
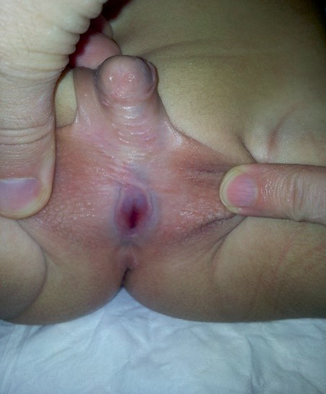
Fig. 66.5
A clinical photograph of a patient with severe hypospadias. This should be investigated to rule out associated DSD
◦ Hypospadias associated with separation of scrotal sacs or undescended testis suggests a DSD.
◦ If the urethral opening is at the base of the phallus, it could be a urogenital sinus in a virilized female.

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


