Disorders of Digestion and Absorption
Ivor D. Hill and Ricardo A. Caicedo
EVALUATION OF THE CHILD WITH MALABSORPTION
The assimilation of ingested nutrients involves a complex integrated process of digestion and absorption with subsequent transport of the breakdown products across the intestinal mucosa into the systemic circulation. Normal digestion and absorption is discussed in Chapter 381. The term malabsorption is broadly used to characterize abnormalities of both digestion and absorption. The schema in Table 408-1 lists the major pathogenic mechanisms of various specific malabsorptive disorders that are generally due to disorders in luminal digestion, mucosal function, or lymphatic transport. Malabsorption may involve multiple nutrients (as occurs with celiac disease or pancreatic insufficiency) or only a single molecule (as found in isolated glucose-galactose malabsorption or vitamin B12 malabsorption). Defects may be congenital, with onset of symptoms shortly after birth, or acquired, when the age of onset of symptoms is variable.
 CLINICAL FEATURES
CLINICAL FEATURES
The evaluation of children with chronic diarrhea is discussed in Chapter 385. The most prominent clinical manifestation of children with disorders of digestion or absorption is diarrhea, except in those conditions involving malabsorption of a single molecule such as vitamin B12. Diarrhea is often accompanied by symptoms of abdominal distension, excessive flatulence, and growth failure in the older infant, toddler, or child. In the newborn or in early infancy, growth failure may not be evident and diarrhea may lead to dehydration. Very watery stools may soak into the diaper and be mistaken for urine, especially in infants with disorders such as congenital chloride-losing diarrhea.1 Stools that are loose to watery in nature and passed in an explosive fashion with excess flatus suggest some form of carbohydrate malabsorption. Carbohydrate-containing stools are generally acidic and frequently excoriate the infant’s buttocks. Large volume or bulky loose stools that are pale and very malodorous characterize steator-rhea, or fat malabsorption, as occurs with pancreatic exocrine enzyme insufficiency or celiac disease.
The time of onset of diarrhea or growth failure may provide clues regarding the diagnosis. Diarrhea or poor weight gain that starts shortly after birth suggests a congenital as opposed to an acquired defect. Onset of symptoms associated with dietary changes may suggest a diagnosis. For example, with sucrase-isomaltase deficiency symptoms commence following ingestion of sucrose-containing formula or foods; in celiac disease, symptoms often follow the introduction of solid, gluten-containing foods into the infant’s diet.2 Exposure to potential pathogens may suggest an infectious etiology of new onset malabsorption.
Previous abdominal surgery can be associated with malabsorption due to short-bowel syndrome, a blind loop, or terminal ileum resection, or malabsorption following partial or complete gastrectomy. Chronic respiratory illnesses occur in cystic fibrosis, which can also cause malabsorption. Arthritis or arthralgia can accompany inflammatory disorders such as Crohn disease. A history of multiple infections raises the possibility of immune deficiency such as Shwachman-Diamond syndrome or HIV.
Growth failure secondary to malabsorption of nutrients usually affects weight first and only impacts linear growth and head circumference at a later stage. Physical examination may show evidence of specific nutrient deficiencies such as pallor (anemia iron deficiency or B12 deficiency), hyperkeratosis (vitamin A deficiency), bruising (vitamin K or C deficiency), glossitis (riboflavin deficiency), dermatitis (niacin or zinc deficiency), rickets (vitamin D and calcium deficiency), muscle wasting with edema (protein deficiency), or peripheral neuropathy (deficiency of vitamin B1, B6, B12). The abdominal examination should include inspection for distension (due to carbohydrate malabsorption and gas production) as well as careful palpation for abdominal masses (tuberculous nodes, lymphoma, neuroblastoma) that may also cause abdominal distension and diarrhea, and for organomegaly. Examination of other systems, such as the respiratory, cardiovascular, and endocrine, is an integral part of the evaluation.
 DIAGNOSTIC EVALUATION
DIAGNOSTIC EVALUATION
The selection of laboratory studies to confirm the presence of malabsorption and establish an etiology will depend upon the most likely causes determined by the history and physical examination. If the history suggests a specific diagnosis such as cystic fibrosis, targeted testing is appropriate. When malabsorption is suspected due to growth failure and diarrhea, screening laboratories guide the subsequent choice of more specialized testing as shown in Table 408-2.3 A complete blood count and differential with smear looking for anemia (iron, folate, or B12 deficiency), acanthocytosis (abetalipoproteinemia), eosinophilia (eosinophilic gastroenteropathy), or lymphopenia (lymphangiectasia) is useful. Measurement of serum bilirubin, alanine aminotransferase, and gamma-glutamyl transpeptidase may identify liver disorders. Low albumin can be due to malnutrition or lymphatic disease. Serum triglycerides and cholesterol may be abnormal in various disorders associated with malabsorption. In most situations serum testing for celiac disease is performed as part of the initial evaluation (see below). An examination of stool for occult blood, fecal leukocytes, or lactoferrin to exclude inflammatory disorders; stool microscopy for parasites such as Giardia; measurement of stool pH and reducing substances to look for carbohydrate malabsorption; and a qualitative test for fecal fat globules (Sudan stain) to look for fat malabsorption may provide valuable information.4 Fecal elastase or chymotrypsin testing provides a useful screening evaluation of pancreatic function, and fecal α1-anti-trypsin can identify protein-losing enteropathies. Sweat tests or genetic testing for cystic fibrosis is usually indicated. If the initial screening tests are suggestive of a malabsorption disorder, more specialized tests are usually required to identify the specific etiology.
Table 408-1. Mechanisms of Maldigestion and Malabsorption
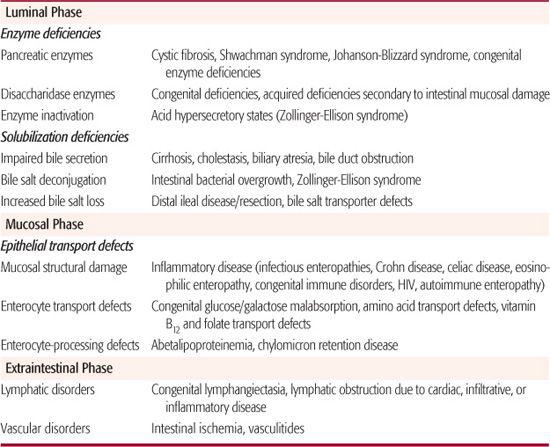
Table 408-2. Diagnostic Studies in the Evaluation of Suspected Malabsorption

In some instances radiologic imaging studies may help identify a specific cause for malabsorption. Barium contrast studies or computerized tomography can identify distal ileal disease as found with Crohn disease, intestinal lymphoma, or tuberculosis and are also useful for identifying malrotations, duplications, blind loops, intestinal strictures, or entero-enteral fistulas that may cause small bowel bacterial overgrowth. Computerized tomography and magnetic resonance imaging may identify abnormalities of the pancreas, including hypoplasia with lipomatous infiltration, intraductal or parenchymal calcifications, or fibrosis, as may occur in chronic pancreatitis.
Tests for Carbohydrate Malabsorption
Malabsorption of carbohydrates is suggested when stool pH is reduced or reducing substances are abnormal. Specific disaccharidase deficiencies can be identified using a breath test that measure hydrogen levels following ingestion of a specific carbohydrate (eg, lactose, sucrose, or fructose). Malabsorption of the carbohydrate is shown by a breath hydrogen level increase of more that 20 ppm due to metabolism of the nonabsorbed carbohydrate by colonic bacteria. Early rises in breath hydrogen suggest possible bacterial overgrowth of the small intestine, whereas a lack of rise may be associated with a lack of hydrogen-producing bacteria due to recent antibiotic usage or to a predominance of methane-producing bacteria. Malabsorption of multiple carbohydrates suggests a decrease in intestinal absorptive capacity as occurs with destruction of the intestinal mucosa. Definitive identification of a disaccharidase deficiency (eg, lactase, sucrase, or glucoamylase) requires endoscopic biopsy, usually from the second or third portion of the duodenum. Pathology reveals normal mucosa, but specific enzyme assays show reduced or absent mucosal enzyme concentrations. The D-xylose test was previously used to assess the integrity of the mucosal surface, but a lack of specificity and sensitivity has the frequent use of this test in contemporary practice.5
Tests for Fat Malabsorption
Low serum levels of vitamins A, D, E, and carotene6 provide indirect evidence for possible fat malabsorption in the child. Further evidence may be obtained through the qualitative determination of fecal fat on a single stool sample using the Sudan stain or by means of the acid steatocrit, which employs a simple, rapid gravimetric method to determine fat content in a small stool sample. This method is reasonably sensitive and specific,7 as are breath tests using carbon 14 triolein as a substrate.8 Definitive quantitative evaluation of fat absorption is unpleasant and inconvenient and is rarely performed in contemporary practice.3 Deficiencies in pancreatic enzymes cause fat mal-digestion.9 The approach to evaluation for these disorders is discussed in Chapter 417. Fat malabsorption may also result from conditions associated with inadequate micelle solubilization: decreased biliary production or secretion (cirrhosis, biliary atresia, or bile duct obstruction), congenital absence of bile salt synthesis, deconjugation and dehydroxylation of bile salts secondary to small intestinal bacterial overgrowth, or depletion of bile salts due to excessive loss in the stools. Normally, bile salts are absorbed in the distal ileum and returned to the liver for reconstitution and excretion. Disruption in this enterohepatic cycle with progressive depletion of the bile salt pool may occur in diseases involving the distal ileum, such as Crohn disease, intestinal lymphoma or tuberculosis, following surgical resection of the distal ileum, or in rare cases, due to congenital defects in the ileal bile salt transporter. Direct assay of blood or duodenal bile salts concentrations is sometimes used to identify a deficiency. An indirect measure of bile salt malabsorption is possible using the 75-selenium-homotaurocholic acid-taurine test but because of the technical requirements for this test, it is not commonly used in clinical practice.
Deficiencies in pancreatic enzymes cause fat mal-digestion.9 The approach to evaluation for these disorders is discussed in Chapter 417. Fat malabsorption may also result from conditions associated with inadequate micelle solubilization: decreased biliary production or secretion (cirrhosis, biliary atresia, or bile duct obstruction), congenital absence of bile salt synthesis, deconjugation and dehydroxylation of bile salts secondary to small intestinal bacterial overgrowth, or depletion of bile salts due to excessive loss in the stools. Normally, bile salts are absorbed in the distal ileum and returned to the liver for reconstitution and excretion. Disruption in this enterohepatic cycle with progressive depletion of the bile salt pool may occur in diseases involving the distal ileum, such as Crohn disease, intestinal lymphoma or tuberculosis, following surgical resection of the distal ileum, or in rare cases, due to congenital defects in the ileal bile salt transporter. Direct assay of blood or duodenal bile salts concentrations is sometimes used to identify a deficiency. An indirect measure of bile salt malabsorption is possible using the 75-selenium-homotaurocholic acid-taurine test but because of the technical requirements for this test, it is not commonly used in clinical practice.
Evaluation of Small Bowel Mucosa
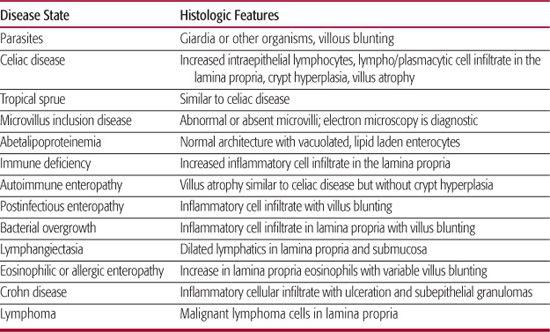
Endoscopic examination of the upper small intestine and colon may identify obvious structural or morphological abnormalities. In most cases, mucosal biopsy is required to identify a specific cause for malabsorption. Routine histologic and, in some cases, electron microscopy examination of small intestinal mucosa is critical for accurate diagnosis. Conditions that may be identified on histology or electron microscopy include celiac disease, protein-sensitive enteropathy, Crohn disease, abetalipoproteinemia, and eosinophilic infiltration. Intestinal biopsies are best performed by means of gastrointestinal endoscopy with multiple biopsies, even when the macroscopic appearance of the mucosa is normal. Table 408-3 lists conditions causing malabsorption that may be identified on small intestinal biopsy.
Additional specialized tests that may be ordered based on the index of clinical suspicion, and initial screening tests include the Schilling test for vitamin B12 malabsorption, and a fasting serum gastrin level and maximal acid output test for Zollinger-Ellison syndrome. These conditions are uncommon in children.
Table 408-3 Histologic Features of Intestinal Diseases That Can Cause Malabsorption
 TREATMENT
TREATMENT
In most cases a specific cause for maldigestion and malabsorption in children can be identified through the process of initial screening tests followed by targeted definitive testing, and treatment addresses the underlying cause. In some cases simple removal of the offending agent is curative, for example, lactose elimination for lactose malabsorption and a gluten-free diet that eliminates all products containing wheat, barley, and rye for celiac disease. In others, enzyme replacement of deficiencies is possible, for example, pancreatic enzyme supplements for cystic fibrosis. In addition to treating the underlying cause, attention should also be focused on correcting any macronutrient and micronutrient deficits that exist at the time of diagnosis in order to promote optimal growth and development of the individual. Periodic assessment of nutritional status and review of nutrient intake is mandatory in all patients with malabsorption syndromes.
CELIAC DISEASE
Celiac disease is defined as a lifelong sensitivity to gluten found in wheat and related proteins found in barley and rye. It occurs in genetically predisposed individuals and is manifest as an immune-mediated enteropathy as defined by characteristic changes seen on small intestinal histology.2 The intestinal mucosal injury leads to varying degrees of malabsorption of nutrients. Removal of wheat gluten and related proteins from the diet usually leads to full clinical remission and restoration of the small intestinal mucosal damage to normality. The fascinating historical aspects of this disorder are found on the DVD. 
 EPIDEMIOLOGY AND ASSOCIATED DISORDERS
EPIDEMIOLOGY AND ASSOCIATED DISORDERS
Celiac disease is one of the most common chronic conditions affecting humans. A prevalence of 1:67 to 1:285 is reported in the European general population.11 A multicenter study involving a large number of individuals in the United States estimates a prevalence of 1:133.12 Celiac disease is diagnosed in North and South America, North Africa, the Middle East countries, India, Australia, and New Zealand, but is rare or nonexistent in black African, Japanese, and Chinese people. The reasons for this are unclear, but could be due to the lack of genetic potential in these populations or to the fact that wheat is not the staple diet in these regions.
 PATHOPHYSIOLOGY AND GENETICS
PATHOPHYSIOLOGY AND GENETICS
There is a strong genetic predisposition for celiac disease. It occurs in 10% to 20% of first-degree relatives, and there is concordance in 30% to 40% of HLA-identical siblings and 70% of homozygous twins.13 Celiac disease is strongly associated with the HLA-DQ2 molecule, which is present in over 90% of all individuals with confirmed disease.13 Most individuals with celiac disease who do not possess the HLA-DQ2 molecule carry the HLADQ8 molecule. While HLA-DQ2 is strongly associated with celiac disease, it is also found in 30% of the general population, hence it is not specific for celiac disease. Other non-HLA genes are believed to play a role in celiac disease, but these have yet to be identified.
Most individuals with celiac disease who do not possess the HLA-DQ2 molecule carry the HLADQ8 molecule. While HLA-DQ2 is strongly associated with celiac disease, it is also found in 30% of the general population, hence it is not specific for celiac disease. Other non-HLA genes are believed to play a role in celiac disease, but these have yet to be identified. 
Dietary Factors
Proteins derived from wheat, barley, and rye initiate the immune processes that result in the intestinal damage found in celiac disease. Although not strictly correct, the term gluten is often loosely used to refer to all proteins found in wheat, barley, and rye. Previously, oats were also implicated, but this now seems to have been due to contamination of oats with wheat flour. There is now good evidence that oats can be safely ingested by the majority of patients with celiac disease.
Additional trigger factors, including viral infections and stress-related events, have been implicated in the onset of disease, but have yet to be conclusively confirmed.
Immunologic Factors
Individuals with untreated celiac disease have increased intestinal permeability rendering their intestinal mucosa more permeable to the passage of harmful peptides. This increase may be related to an upregulation in zonulin that has been found in individuals with celiac disease.17 Zonulin is a protein that exerts an influence on the enterocyte microskeleton to control the tight junctions in the intestinal epithelial layer. Passage of certain peptides from gluten and related proteins across the enterocyte layer triggers the innate response in susceptible individuals that produces a lymphocytic cytotoxic reaction against enterocytes. 
In individuals with celiac disease, the gluten-derived peptides that traverse the intestinal mucosa are targeted by tissue transglutaminase, and glutamine molecules are deamidated to glutamic acid. This results in a great increase in the negative charge on the peptide being greatly enhanced and binding to the peptide groove on the DQ2 molecule of the antigen presenting cells is increased. The bound peptide antigen is presented to T lymphocytes, which in turn stimulate B lymphocytes to elaborate a number of antibodies to gliadin, endomysium, and tissue transglutaminase. T lymphocytes also elaborate additional cytokines, including IFN-α, which are believed to play a role in the mucosal remodeling that occurs in celiac disease.19
 CLINICAL FEATURES AND DIFFERENTIAL DIAGNOSIS
CLINICAL FEATURES AND DIFFERENTIAL DIAGNOSIS
The manifestations of celiac disease are protean with tremendous variation among affected individuals (Table 408-4).20 The “classic” constellation of symptoms composing malabsorptive-type diarrhea, poor weight gain, abdominal distension, and proximal muscle wasting in the child under 2 years of age is readily recognized as a manifestation of celiac disease. Today, this type of presentation accounts for a relatively small number of all newly diagnosed cases. In rare cases, very young children present with a celiac crisis characterized by severe diarrhea with electrolyte imbalance, hypoalbuminemia, and a shocklike state that can be fatal.
Table 408-4. Clinical Manifestations of Celiac Disease
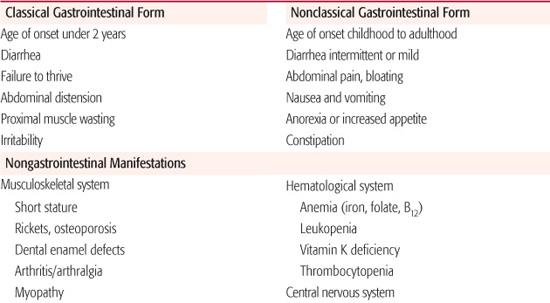
More commonly, symptoms of celiac disease are delayed until late childhood, adolescence, or even adulthood. Population-screening studies using serologic tests to identify those who might have celiac disease have led to the recognition that many individuals with characteristic changes seen by small intestinal biopsy are asymptomatic, indicating that the intestinal mucosal damage likely may precede onset of symptoms by many years. Most symptomatic children will have gastrointestinal symptoms as their initial manifestation of celiac disease. Diarrhea is frequently present, but may be mild or intermittent for prolonged periods of time. Abdominal pain, distension, and a feeling of bloating with flatulence are fairly frequent complaints in the older child and adolescent and may be incorrectly attributed to the irritable bowel syndrome or lactose intolerance. Nausea and vomiting occur less commonly, and in some cases, constipation has been the major complaint. In adolescents and adults, symptoms of celiac disease may be indistinguishable from those of irritable bowel syndrome, resulting in the delayed diagnosis of celiac disease in many individuals.
Nongastrointestinal Manifestations
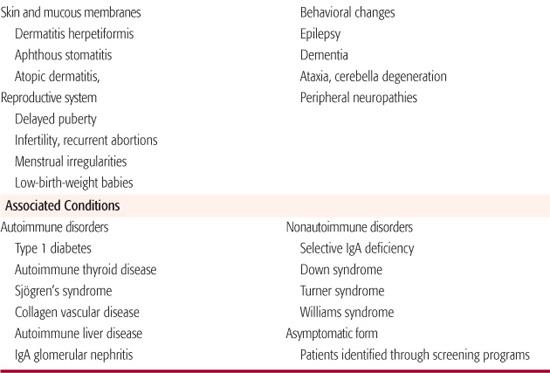
Nongastrointestinal manifestations of celiac disease are being recognized with greater frequency, and in adults these may account for half of all newly diagnosed cases (Table 408-4). Decrease in linear growth velocity or delayed onset of puberty may be the initial manifestation of celiac disease in the pediatric population. Dental enamel hypoplasia, involving all 4 quadrants of the secondary dentition in a symmetrical distribution, is well described in celiac disease and may be the initial manifestation first recognized during a routine dental examination. Dermatitis herpetiformis, characterized by extremely itchy bullous lesions on the extensor surfaces of the limbs, trunk, and scalp, is a feature of celiac disease that is more common in adults and can occur without any intestinal symptoms (Fig. 408-1). Behavioral changes, fatigue, anorexia, and unexplained iron-deficiency anemia are additional initial manifestations.
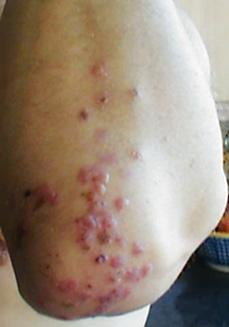
FIGURE 408-1. Celiac disease presenting as dermatitis herpetiformis.
Associated Conditions
Celiac disease is also strongly associated with a number of autoimmune conditions (Table 408-4).10,20 Up to 10% of patients with type 1 diabetes have been found to have celiac disease, and the condition occurs in about 12% of those with Down syndrome, leading to recommendations for routine screening in these and other high-prevalence populations.10 Selective IgA deficiency occurs in about 3% of patients with celiac disease as compared to about 1:500 of the general population, complicating the diagnosis of celiac disease with anti-IgA antibody–based diagnostic test.
 DIAGNOSTIC EVALUATION
DIAGNOSTIC EVALUATION
Revised criteria for the diagnosis of celiac disease published by the European Society of Pediatric Gastroenterology and Nutrition in 199021,22 and endorsed by the North American Society of Pediatric Gastroenterology and Nutrition in 200510 list 2 mandatory requirements for the diagnosis of celiac disease: (1) identification of the characteristic histologic findings on small intestinal mucosal histology in a symptomatic patient and (2) complete symptom resolution on a strict gluten-free diet. The use of serologic tests may be used as supportive evidence for the diagnosis, but despite the improved sensitivity and specificity of new serologic tests, serology alone is considered inadequate for initiation of lifetime therapy that has substantial social and economic consequences.
Serologic tests are useful for screening of patients with possible celiac disease or high-risk groups that may have asymptomatic disease. Commercial laboratories offer a number of serologic tests for celiac disease. Those most readily available include the antigliadin antibodies (AGA), antiendomysium antibodies, and anti-tissue transglutaminase antibodies. The AGA tests have highly variable sensitivities and specificities23,24 and are no longer recommended for routine use as a screening test. The antiendomysium antibody test (EMA) and anti-tissue transglutaminase antibody test (TTG) are both highly sensitive (> 95%) and specific (> 98%) and are recommended as the test of choice for screening purposes. Current recommendations from the North American Society for Pediatric Gastroenterology, Hepatology and Nutrition, the American Gastroenterology Association, and the NIH advocate use of either the TTG or EMA for routine screening of patients suspected of having celiac disease10,25,26 or in patients at risk of celiac disease (Fig. 408-2). Serum IgA level is recommended to be obtained at the time of testing to identify those who are IgA deficient since IgA-based serologic tests are of no use in such individuals, for whom IgG-based tests for EMA or TTG should be considered. EMA and TTG tests are also less reliable in children less than 2 years of age,23 so in this age group AGA tests combined with the TTG or EMA may increase sensitivity during screening, although if the diagnosis is likely, small bowel biopsy may be indicated.
Small intestinal biopsy remains the definitive test for the diagnosis of celiac disease.10 Intestinal biopsy specimens are usually obtained by means of an upper gastrointestinal endoscopy. Visual abnormalities of the mucosa may be recognized at endoscopy (Fig. 408-3). These include mucosal nodularity, scalloping along the borders of the folds, and a mosaic pattern. These changes have been shown to correlate with the degree of mucosal atrophy, but have not been shown to have adequate sensitivity or specificity for diagnosis. For these reasons, even if the endoscopic appearance is completely normal, multiple samples should be obtained from the second part of the duodenum or beyond. Characteristic histologic features of celiac disease are progressive and include an increase in the number of intraepithelial lymphocytes, an increase in plasma cells in the lamina propria, loss of enterocyte basal nuclear polarity, crypt hyperplasia with an increased crypt mitotic index, and villous blunting with reversal of the villous height-to-crypt depth ratio (Fig. 408-4).19 Disease severity is characterized by pathologists using the Marsh classification.27 Other disorders such as autoimmune enteropathy and milk protein–sensitive enteropathy may cause flattening of the intestinal mucosa together with a mucosal inflammatory cell infiltrate that could be mistaken for celiac disease.
Other disorders such as autoimmune enteropathy and milk protein–sensitive enteropathy may cause flattening of the intestinal mucosa together with a mucosal inflammatory cell infiltrate that could be mistaken for celiac disease.
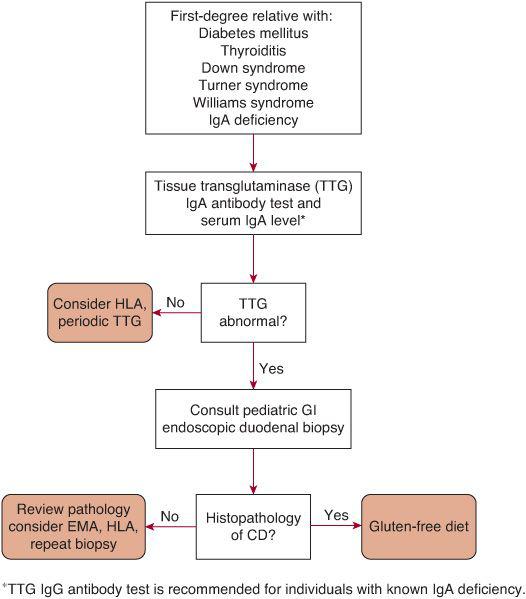
FIGURE 408-2. Management of children in at-risk groups for celiac disease. Patients that do not possess the HLA-DQ2 or HLA-DQ8 molecule are at low risk. CD, celiac disease; EMA, endomysium antibody; HLA, HLA typing.
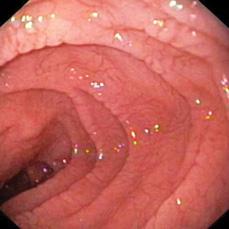
FIGURE 408-3. Endoscopic findings of scalloping of the mucosal folds in celiac disease.
 TREATMENT
TREATMENT
The treatment approach to celiac disease is summarized in eFigure 408.1  . A diagnosis of celiac disease mandates lifelong dietary exclusion of gluten found in wheat and related peptides in barley and rye. Previously, oats were excluded, but this does not appear to be necessary if the oats are milled separately from gluten-containing grains. Strict adherence to such a diet is difficult and requires constant vigilance on the part of the patient. For this reason, it is essential to confirm the diagnosis by means of an intestinal biopsy before starting an exclusionary diet. In the short term, dietary modification will result in rapid remission of symptoms and correction of growth and nutritional deficiencies.
. A diagnosis of celiac disease mandates lifelong dietary exclusion of gluten found in wheat and related peptides in barley and rye. Previously, oats were excluded, but this does not appear to be necessary if the oats are milled separately from gluten-containing grains. Strict adherence to such a diet is difficult and requires constant vigilance on the part of the patient. For this reason, it is essential to confirm the diagnosis by means of an intestinal biopsy before starting an exclusionary diet. In the short term, dietary modification will result in rapid remission of symptoms and correction of growth and nutritional deficiencies.
Long-term benefits of adhering to the diet include preventing such complications as osteoporosis and reducing the risk for intestinal malignancies. Repeating the serologic tests at intervals may help the physician and patient monitor whether gluten and related products are being adequately excluded from the diet. Persistent elevation of a serologic marker, or recurrence of a positive test that had previously been negative, is indicative that the patient is knowingly, or unknowingly, continuing to ingest gluten. Elevation of serologic antibody levels and recurrence of significant small intestinal mucosal damage after initial recovery can precede symptoms by several years in those who do not strictly adhere to the diet.
Adherence to the diet can be particularly difficult in children, and consultation with an experienced pediatric dietician is recommended. Cookbooks and various gluten-free specialty items are available for patients with celiac disease and are easily accessed via the Internet, but strict adherence to the diet is challenging for those with limited resources. The American Dietetic Association has published evidence-based guidelines for treatment of celiac disease.28,29 Patients with celiac disease are also encouraged to join a local support group, as these provide valuable sources of education and dietary guidance. Children should see their physician on a regular basis to monitor their growth and general health. At these visits, the physician should take the opportunity to evaluate the patient’s adherence to the gluten-free diet and provide ongoing education on the long-term benefits of dietary compliance.30
Current research is aimed at finding alternative forms of therapy for patients with celiac disease. One possibility that is being explored is the use of bacterial prolyl endopeptidases with food to further digest the potentially harmful gluten peptides and render them nontoxic. Other possible forms of therapy involve the use of immune modulators to block specific inflammatory mediators that are involved in the immunopathogenesis of the disease. The safety and efficacy of these alternative forms of therapy remain to be proven.
 COMPLICATIONS
COMPLICATIONS
Untreated celiac disease has potential for long-term adverse health consequences.28 Mortality is increased twofold over the general population in those with symptomatic celiac disease who are noncompliant with therapy. Most of the excess deaths in these cases are related to malignancies, particularly intestinal lymphomas. Individuals with celiac disease are at significantly increased risk for intestinal lymphomas and in particular an enteropathy-associated T-cell lymphoma that carries a poor prognosis. In those who remain compliant with dietary therapy, the relative risk for intestinal cancers returns to that of the general population after a period of 5 years. In children with growth stunting due to celiac disease, early diagnosis and treatment usually results in catchup growth. However, if the diagnosis and treatment is delayed beyond puberty, there may be permanent growth stunting, with the child failing to reach his or her true growth potential. A decrease in bone mineralization leading to osteopenia or frank osteoporosis is frequently present at the time of diagnosis in those with symptomatic celiac disease. In children, early diagnosis and treatment of celiac disease results in restoration of normal bone mineralization. In adults, treatment will improve osteopenia or osteoporosis in most cases, but bone mineralization seldom returns to that of healthy individuals without celiac disease. It has also been suggested that a delay in diagnosis and treatment of celiac disease may place the individual at increased risk for developing additional autoimmune disorders such as type 1 diabetes and autoimmune thyroiditis. However, the evidence supporting the concept that prolonged exposure to gluten is a cause for other autoimmune disorders is conflicting.
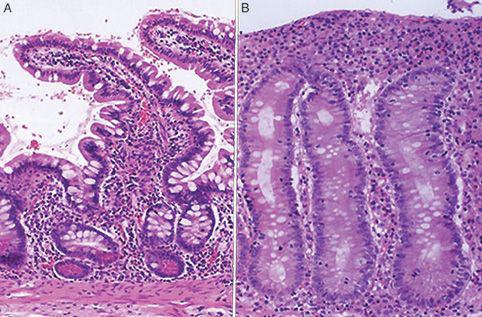
FIGURE 408-4. A: Normal small intestinal biopsy. B: Celiac disease showing villous inflammation.
IDIOPATHIC DIARRHEA OF INFANCY
Chronic idiopathic diarrhea of infancy is also known as intractable diarrhea of infancy, protracted diarrhea of infancy, severe persistent diarrhea of infancy, and prolonged diarrhea of infancy.31 A number of new disorders that cause diarrhea during infancy have been recognized in recent years,32-35 but despite these advances there remains a group of infants for whom no specific etiology can be identified. The term chronic idiopathic diarrhea of infancy remains appropriate for these cases. In the past, this term was applied to infants less than 3 months of age, but it now is often used to describe any infant with otherwise unexplained chronic diarrhea, characterized by ongoing watery diarrhea lasting for more than 2 weeks. Stool losses often exceed 30 mL/kg/day. The volume of stool loss is usually of such magnitude that the infant is unable to maintain adequate hydration without additional intravenous fluids. Without appropriate management, mortality rates are exceedingly high, thus early recognition of the disorder is important to improve outcome.
 EPIDEMIOLOGY AND PATHOPHYSIOLOGY
EPIDEMIOLOGY AND PATHOPHYSIOLOGY
The commencement of diarrhea in this disorder resembles that in an acute infectious episode of either viral or bacterial gastroenteritis. Apparent risk factors for persistent diarrhea in an infant following such an illness include prematurity, early age of onset of the diarrhea, lack of breast-feeding, low socioeconomic circumstances with poor hygiene, and preexisting malnutrition. In most cases an inciting infectious agent is not identified, but even when a bacterial agent is identified and treatment administered, diarrhea may persist. Small intestinal mucosal biopsy of these infants consistently demonstrates subtotal to total villous atrophy, with an increased cellular infiltrate and cuboidal epithelial cells. It appears that in these infants villous repair is ineffective. Histologic evidence of intestinal mucosal injury can persist for several months following the initial clinical presentation, even in those who demonstrate clinical recovery from the diarrhea.36 The intestinal villous atrophy results in deficiencies of intestinal brush border enzymes. This leads to secondary disaccharide (and sometimes even monosaccharide) intolerance. Enterocyte destruction may also reduce enteric hormone secretion, resulting in a secondary pancreatic insufficiency that aggravates the malabsorption of fats, carbohydrates, and proteins. Nutrient absorption decreases to such an extent that mucosal healing is impaired due to a lack of nutrients, resulting in a spiraling downward course.37 Many of these infants also develop small intestinal bacterial overgrowth, possibly due to hypochlorhydria, which may further damage the intestinal mucosa and can deconjugate bile acids, causing increased fluid and electrolyte losses by the colon.38,39
 CLINICAL FEATURES AND DIFFERENTIAL DIAGNOSIS
CLINICAL FEATURES AND DIFFERENTIAL DIAGNOSIS
The diagnosis of idiopathic diarrhea of infancy is made by exclusion of other known causes of chronic diarrhea in an infant with a typical history and presentation.40 Conditions that cause severe ongoing diarrhea in the very young infant need to be excluded: enteropathies associated with the villous atrophy and transporter defects (see Table 408-5), pancreatic insufficiency (see Table 417-1) malrotation and Hirschsprung disease, and rarely, endocrinopathies such as those associated with hypoparathyroidism, hyperthyroidism, adrenal insufficiency, VIPomas, and neuroblastoma.32-35 Diarrhea that starts within the first few days of life should always raise suspicion for one of the congenital structural, metabolic, mucosal transport, or enzyme abnormalities. A preceding history of polyhydramnios in the mother may be a clue to the diagnosis of congenital sodium- or congenital chloride-losing diarrhea in the infant. Diarrhea that starts some months after birth is more likely to be due to an acquired disorder. Chronic diarrhea starting only after the introduction of solid foods may be indicative of celiac disease in the older infant, and, although very rare in the young infant, both Crohn disease and ulcerative colitis have been described.
Because of the complexity involved in reaching a final diagnosis and the potential high mortality associated with idiopathic diarrhea of infancy, infants with severe, persistent diarrhea should be referred to a center with the facilities and expertise necessary for investigation and treatment. Initial laboratory evaluation generally includes determination of serum electrolytes and acid-base status; stool microscopy for leukocytes, ova, and parasites; repeated bacterial culture; and reducing substances. Measurement of stool electrolyte content is often helpful to confirm or exclude a secretory-type diarrhea, which is characteristic of some congenital conditions causing chronic diarrhea. Differentiating a secretory-type diarrhea from that which is osmotically induced can also be determined on a clinical basis by temporarily withholding feeds from the infant and providing all fluids via the intravenous route. Stool output will persist unabated in those with a secretory-type diarrhea, whereas it will decrease dramatically in those with osmotically induced diarrhea, only to recur with reintroduction of feedings. Evaluation for other causes of chronic diarrhea, including infection, immunodeficiency, and pancreatic insufficiency, may be considered.
An intestinal biopsy is not always necessary to make a presumptive diagnosis and to initiate treatment. When a biopsy is obtained, it is used to rule out other diagnosis and should include specimens for both light and electron microscopy, as well as for Sudan fat and neurogenin-3 staining. Biopsy confirms findings of villous atrophy, but the cause of atrophy is nonspecific.
 TREATMENT AND PROGNOSIS
TREATMENT AND PROGNOSIS
Preventing the evolution of acute infectious diarrheal episodes into chronic “idiopathic diarrhea” should be a primary goal for infants who are at risk for this condition. Strategies that are of benefit in this regard include promoting breast-feeding in all cases and promoting continued feeding of children with acute diarrhea to prevent any deterioration in their nutritional status. In infants with persistent diarrhea, the first priority is to prevent dehydration and to correct electrolyte imbalance as discussed in Chapter 385. If diarrhea persists when enteral feeding ceases, a diagnostic workup should begin as discussed above. If diarrhea resolves, attempts to enterally feed should resume.
Often infants can tolerate smaller-volume feeds, particularly if delivered as a continuous nasogastric feeding. Modular formula with decreased carbohydrate or semielemental formula are often better tolerated than lactose-containing formula. If adequate calories can be delivered without substantial increases in stool volume and without dehydration, the enteral approach is preferable. If stool volume increases substantially as enteral feeding is attempted, the enteral feeding rate should be reduced to a smaller amount that is better tolerated and parenteral nutrition should be administered to provide additional nutritional support.
Usually, within 5 to 7 days of starting parenteral nutrition the infant will tolerate small-volume feeds of a reduced-carbohydrate modular formula, a semielemental, or elemental formula without any marked increase in stool output. The volume of formula feeds can then be slowly increased with simultaneous decrease in the amount of parenteral nutrition. In those infants showing steady improvement as demonstrated by the ability to receive an increasing proportion of calories enterally, expectant management is reasonable. In those infants who do not improve, small bowel biopsy should be performed to rule out other causes of persistent diarrhea.
Empirical treatment of small intestinal bacterial overgrowth with a combination of oral antibiotics (gentamicin 50 mg/kg/day in 6 divided doses for 3 days and metronidazole 15 mg/kg/day in 3 divided doses) and cholestyramine (0.5–1 g every 6 hours for 5 days) has proven highly beneficial and may result in a dramatic decrease in stool output in many cases.39 Underlying vitamin and mineral deficiencies also require correction. Both vitamin A and zinc deficiency are frequently documented in malnourished infants and are associated with prolongation of diarrhea in these cases. Complete recovery is now possible in the majority of infants with chronic idiopathic diarrhea who receive appropriate therapy.
OTHER GENERALIZED MUCOSAL DISORDERS CAUSING MALABSORPTION
There are a number of enteropathies associated with varying degrees of malabsorption of nutrients. In some there is histologic evidence of structural derangement characterized by decreased villus height with consequent loss of intestinal absorptive surface area. In others the mucosal architecture appears intact, and malabsorption results from a functional defect of the enterocyte (Table 408-5). Enteropathies associated with malabsorption can be congenital or acquired. Congenital enteropathies usually present early in infancy, and the underlying defect is generally not reversible. Acquired enteropathies can present at any age and are potentially reversible, depending on the underlying cause.
CONGENITAL ENTEROPATHIES
 MICROVILLUS INCLUSION DISEASE
MICROVILLUS INCLUSION DISEASE
Congenital microvillus inclusion disease has also been referred to as congenital microvillus atrophy and familial microvillus atrophy.41 It is believed to be inherited in an autosomal-recessive manner based on occurrence among siblings and clustering among infants of Navajo descent.42 The entity is characterized by watery diarrhea, with onset within the first days after birth, although about 20% present later over the first 2 months of life. Stool and electrolyte losses are of such magnitude that rapid dehydration and death will ensue unless appropriate treatment with intravenous fluid is initiated. Massive fecal losses (50 to 800 mL/kg/day) continue even when the infant is maintained on parenteral nutrition and given nothing by mouth. The diarrhea is secretory, with increased electrolyte concentrations without an anion gap. Nutrition cannot be maintained via the enteral route, and parenteral nutrition is required. 
Histologically, the small intestine demonstrates diffuse villus atrophy, but without crypt hypertrophy or an inflammatory cell infiltrate. The surface enterocytes are disorganized, with loss of brush border definition, and there is extensive apical cytoplasmic vacuolization (eFig. 408.2  ). Electron microscopy reveals mucosal surface enterocytes that lack microvilli completely or that have sparse, shortened villi. The apical cytoplasm of the enterocyte contains a marked increase in electron-dense secretory granules of various sizes, some of which contain glycocalyx and brush border–related material (Fig. 408-5). The hallmark of the disease is the presence of microvilli within involutions of the apical membrane.
). Electron microscopy reveals mucosal surface enterocytes that lack microvilli completely or that have sparse, shortened villi. The apical cytoplasm of the enterocyte contains a marked increase in electron-dense secretory granules of various sizes, some of which contain glycocalyx and brush border–related material (Fig. 408-5). The hallmark of the disease is the presence of microvilli within involutions of the apical membrane.
Treatment has been largely unsuccessful. In nearly all cases, life can be sustained only by parenteral nutrition unless the infant can undergo intestinal transplantation.
 TUFTING ENTEROPATHY
TUFTING ENTEROPATHY
Tufting enteropathy is also known as intestinal epithelial dysplasia. In some cases the infants may have characteristic dysmorphic facial features.43 The condition is characterized by chronic diarrhea beginning shortly after birth and is associated with poor growth. In some cases nutrition cannot be maintained via the enteral route alone, and additional parenteral nutrition is required for normal growth.
Histology of the small intestine reveals varying degrees of villus atrophy with crypt hyper-plasia and a minimal increase in inflammatory cells in the lamina propria (Fig. 408-6). The characteristic feature is the presence of foci of closely packed enterocytes with rounding of the apical membrane resembling “tufts” that appear to be coming away from the epithelium. Because of the patchy nature of these findings, several intestinal biopsies may be needed in order to make the diagnosis. Biopsies may appear near normal early in life, showing only nonspecific signs of villous atrophy, with characteristic tufts on later biopsy. The disorder appears to be due to an abnormality of the basement membrane or cell-matrix interactions. Mice in which the gene encoding the transcription factor Elf3 is disrupted have morphologic features resembling tufting enteropathy.44
The condition is believed to be inherited in an autosomal recessive manner and is more common in those of Arab descent.45 The long-term prognosis is variable. A small number of affected individuals receive adequate nutrition via the enteral route, but most cases require parenteral nutrition. There is a report of a successful pregnancy in a woman with tufting enteropathy.46 In those cases requiring long-term supplemental parenteral nutrition, the only other therapeutic option is intestinal transplantation.
Table 408-5. Enteropathies Causing Malabsorption



