14 Urologic Disorders
Physical Examination
Examination begins with a general overview of the patient. Hemihypertrophy, congenital scoliosis, an abnormal gait, facial or external ear deformities, or the presence of multiple congenital anomalies may be associated with urologic disorders. Abdominal examination should begin with inspection for visible masses, followed by gentle palpation. Enlarged kidneys are usually palpable as upper abdominal or flank masses in small children and infants, but may be difficult to appreciate in older children. An enlarged bladder or lesion of gynecologic origin may be palpable as a midline mass arising out of the pelvis. Abdominal masses should be characterized as cystic or solid; smooth, lobulated, or irregular; fixed or mobile; and tender or nontender. The abdominal portion of an abdominoscrotal hydrocele may be palpable in the left or right lower quadrant, and may increase in size when the scrotal component (large hydrocele) is compressed (Fig. 14-1).
The inguinal areas should be examined for palpable gonads. Gonads palpable in the groin are frequently quite mobile and may move during examination from the inguinal canal almost into the scrotum, or lateral to the scrotum toward the perineum. They may also move from a palpable position in the groin to disappear into the abdomen during examination; therefore high testes may be palpable on one examination and nonpalpable on the next. The sensitivity of palpation for detection of inguinal testes and other masses may be increased by the use of soap or lubricant on the examiner’s fingers. It may be difficult to palpate an inguinal testis by trying to “pinch” it between the fingers, whereas lubricated fingers gliding over the inguinal canal may easily detect a gonad as it slides beneath the fingers. The best technique is to place the fingers flat, over the inguinal canal and above the level of the internal inguinal ring, and slide them slowly toward the pubis, then over the pubis toward the scrotum and down over the perineum lateral to the upper scrotum. On occasion, a vas and spermatic cord palpable in the groin (most notably where it passes over the pubic tubercle) may end in a small “nubbin” (remnant of an atrophic testis), which may be palpated in the upper scrotum or just above the scrotum. The structures of the inguinal canal should be examined in a similar fashion to detect thickening of the cord structures that may be seen in the presence of an inguinal hernia or communicating hydrocele. Masses in the inguinal canal may also represent an inguinal hernia containing bowel, omentum, or bladder (or ovary in girls, perhaps a testis in a phenotypic female with androgen insensitivity) (Fig. 14-2), communicating hydrocele or hydrocele of the cord, or malignancy of the paratesticular tissues or cord structures (i.e., sarcoma). Inguinal lymphadenopathy is usually detected lateral to the inguinal canal.
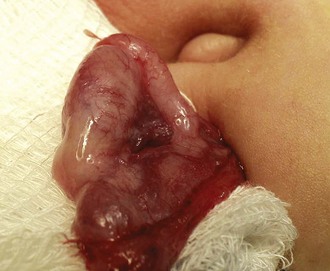
Figure 14-2 Androgen insensitivity. Left inguinal testis in an XY phenotypic female with androgen insensitivity.
Genital examination should include both inspection and palpation. The penis should be of appropriate length and diameter. Penile stretch length can be determined by using a ruler or tongue blade pressed against the pubic symphysis as the penis is gently stretched alongside it and the position of the tip of the glans marked for measurement. A concealed or buried penis may occur after circumcision or may be congenital. In most cases, hidden penises are retractile, in large part due to a thick suprapubic fat pad. Most cases of buried penis will resolve with time, whereas in severe cases the tethering may be due to dysgenetic fascial attachments and will require surgical correction. It may be difficult to determine the difference between the two in younger boys. Penoscrotal fusion or webbing may also cause an anomalous appearance and entrapment of the penis (see Fig. 14-32).
Antenatal Urinary Tract Dilation
Detection of urinary tract dilation (hydronephrosis, hydroureteronephrosis, pyelectasis, pyelocaliectasis) in the fetus is common, either during screening for a fetal anomaly or as a serendipitous finding (Fig. 14-3). All fetal urinary tract dilation demands some degree of postnatal evaluation. Most cases of hydronephrosis, even of a significant degree, are not detectable by physical examination of the neonate. Because only the most severe cases present as an abdominal or flank mass, a distended bladder, or generalized increase in abdominal girth and thus lead to immediate uroradiologic evaluation, most questions surround those infants with modest fetal hydronephrosis and a normal postnatal physical examination. All infants with fetal urinary dilation should have postnatal ultrasonography. In severe bilateral cases, ultrasound may be performed urgently, whereas in less severe hydronephrosis, ultrasound examination may best be performed 3 to 10 days after delivery to allow increased urine production to fill out dilated systems that may be relatively decompressed in the immediate postnatal period, especially in a relatively dehydrated infant. When postnatal ultrasound is normal (spontaneous resolution of fetal hydronephrosis occurs in up to 20% of cases) (Fig. 14-4), follow-up ultrasound should be performed at several months and perhaps at 1 year because delayed reappearance of dilation has been reported.
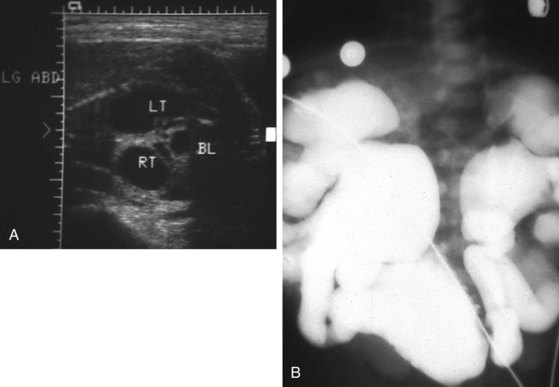
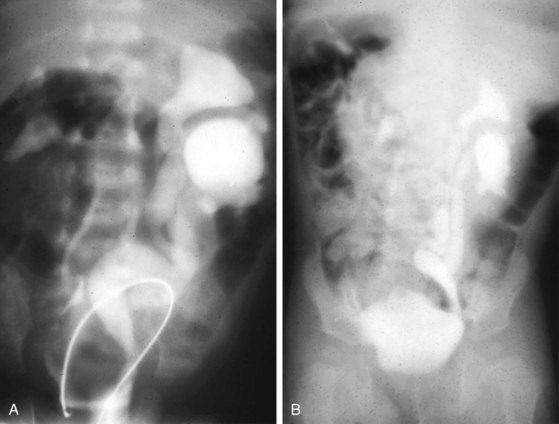
When postnatal hydronephrosis is documented, complete radiographic evaluation is indicated (an exception may be made in very mild cases of pyelectasis in males who may require only limited ultrasound follow-up). If dilation is severe, renal function poor, or coexistent anomalies demand it, evaluation should be performed as soon as possible. Voiding cystourethrography is an integral part of the evaluation of every infant with hydronephrosis and should be the first study performed in all cases to rule out both infravesical obstruction and vesicoureteric reflux (see Fig. 14-3, B). Subsequent evaluation by intravenous urography, radionuclide scan, or both may be appropriate to assess function and to document whether true obstruction or mere dilation is present. If the infant is in stable condition (particularly when the lesion is unilateral or moderate in degree with good renal function), radionuclide evaluation should be delayed for 4 to 6 weeks or longer to allow improved glomerular filtration, making studies more accurate. If nonobstructive dilation is documented, long-term follow-up may document gradual resolution of the hydronephrosis in some cases and persistent dilation in others.
Posterior Urethral Valves
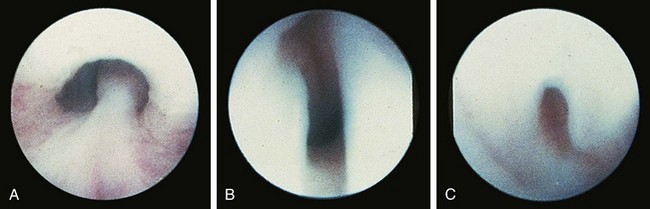
Valvular obstruction of the posterior urethra is a common cause of infravesical obstruction in males. Presentation may be associated with prenatal hydronephrosis (see Fig. 14-3), urinary tract infection, incontinence, or renal failure. Although the classic presentation of infants and older children with urethral valves is a diminished urinary stream, few children are actually evaluated because of this symptom. Neonates may present with severe pulmonary hypoplasia and renal failure or with bladder distention and hydroureteronephrosis. Older boys with unrecognized urethral valves may present with symptoms ranging from incontinence to renal failure. Voiding cystourethrography is the key to the diagnosis of urethral valves. In most cases endoscopic fulguration of urethral valves is undertaken (Fig. 14-5, A-C). In some cases cutaneous vesicostomy may be indicated, especially in tiny babies in whom urethral instrumentation may be problematic. Rarely is upper tract diversion (ureterostomy or pyelostomy) indicated. The ultimate prognosis for renal function depends on the state of the renal parenchyma (whether or not dysplasia is present) and of bladder compliance (poorly compliant bladders with diminished elasticity may fail to allow reflux or hydronephrosis to improve after valve ablation and may be associated with a worse prognosis for continence and renal function).
Cryptorchidism
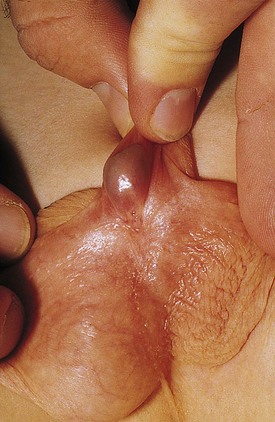
Cryptorchidism occurs in approximately 33% of premature and 3% of full-term boys. Observing gradual testicular descent over several weeks in a premature infant is not unusual. Cryptorchidism is associated with many syndromes but rarely with urinary tract anomalies. The exception is true congenital monorchism, which may be associated with ipsilateral renal agenesis. Renal ultrasound is indicated. In most cases of an absent testis, however, spermatic structures are present, which indicate that the testis was formed but subsequently atrophied due to a prenatal event. In these cases, concern for ipsilateral renal agenesis is minimal. Conversely, ultrasound examination of the lower quadrant and inguinal canal to search for a nonpalpable testis is not indicated and is a highly unreliable examination. Hypospadias associated with even unilateral cryptorchidism should raise the question of intersex anomalies, and karyotype should be determined in the neonate (Fig. 14-6). When bilateral nonpalpable testes are present in infancy, endocrinologic evaluation (serum follicle-stimulating hormone [FSH], luteinizing hormone [LH], testosterone) may determine whether functional testicular tissue exists. The infant with cryptorchidism should be monitored closely, with hormonal or surgical treatment undertaken at about 6 months of age.
Prune-Belly (Eagle-Barrett or Triad) Syndrome
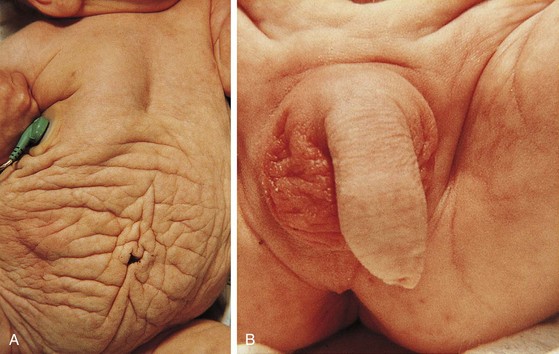
Prune-belly syndrome has a fascinating constellation of physical findings and occurs almost exclusively in boys at a rate of 1 in 35,000 to 50,000 live births. The triad includes abnormal abdominal musculature (variable degrees of muscular laxity, which may be asymmetrical); abdominal cryptorchidism; and floppy dysmorphic urinary tracts, most with vesicoureteric reflux (Fig. 14-7). Plain abdominal radiography demonstrates the bell-shaped thorax and abdomen seen in this syndrome. The typical prune-belly refluxing ureter, with increasing tortuosity in the lower portion, is revealed by voiding cystourethrogram or intravenous urogram. Because of the severe risk of sepsis associated with urinary tract infection, urethral catheterization should be undertaken only with the utmost care to prevent infection and with antibiotic prophylaxis. Megalourethra may be seen. Associated with the syndrome in most patients are prostatic hypoplasia and dimples on the lateral aspects of the knees, which some experts believe are secondary to an exaggerated cross-legged position in utero. Gastrointestinal and cardiac anomalies occur in a proportion of patients, but the factor that most determines longevity is the presence and degree of renal dysplasia.
Anomalies of the Urachus
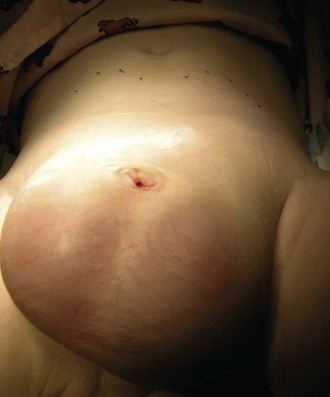
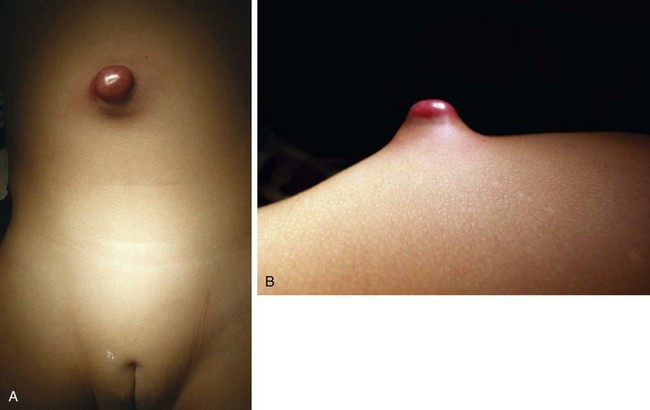
The urachus extends from the bladder dome to the umbilicus and is usually a vestigial structure during extrauterine life. Several lesions may result from persistence of the urachus: patent urachus, vesicourachal diverticulum, urachal cyst, and alternating urachal sinus. Many of these lesions go unrecognized for long periods before becoming symptomatic. Patent urachus results when the urachal lumen fails to obliterate and the bladder communicates with the umbilicus (Fig. 14-8). Umbilical drainage, inflammation, or infection may result. Voiding cystourethrography is important in the evaluation of possible urachal disorders and to exclude infravesical obstruction. The differential diagnosis includes persistent omphalomesenteric duct. Urachal cysts may become infected and present in infancy through adulthood with suprapubic or infraumbilical pain, tenderness, a palpable mass, or abdominal wall inflammation (Fig. 14-9). Urinary tract infection with irritative voiding symptoms or sepsis may result. Ultrasonography or computed tomography (CT) may aid in diagnosis. Urachal diverticula are usually clinically inconsequential unless they are large enough to cause urinary stasis and infection. Because unrecognized urachal remnants may be the source of carcinoma in adults, many cases with a poor prognosis due to delayed detection, even asymptomatic urachal lesions, should be excised. Many “urachal” lesions in newborns turn out to be merely localized umbilical granulomas.

Hydronephrosis
Ureteropelvic Junction Obstruction
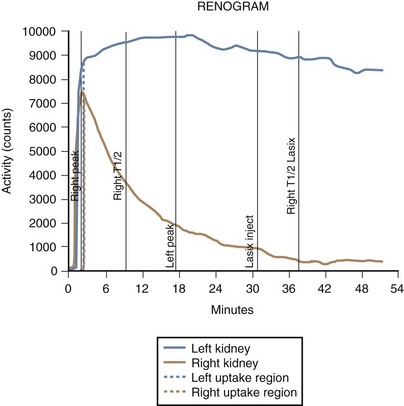
Lesions of the ureteropelvic junction (UPJ) are a common cause of hydronephrosis. UPJ obstruction may present as antenatal hydronephrosis, neonatal flank mass, urinary tract infection, or recurrent abdominal pain in the older child and adolescent. In many cases of significant obstruction the kidney may not be palpably enlarged. UPJ obstruction may be documented by ultrasound or intravenous pyelography and confirmed by retrograde pyelography (Fig. 14-10, A). Voiding cystourethrography is important, particularly in infants, because vesicoureteric reflux may coexist with UPJ obstruction. In some cases reflux is the primary lesion, with the UPJ kink as a secondary lesion (Fig. 14-10, B), whereas in other cases, severe reflux may be the primary lesion with a secondary kink at the ureteropelvic junction, which may or may not prove to be obstructive (Fig. 14-10, C). Not all hydronephrotic kidneys are truly obstructed. In borderline cases, radionuclide diuresis renography (Lasix renal scan) (Fig. 14-11) or percutaneous antegrade pressure perfusion studies (Whitaker test) may be necessary to determine whether surgical intervention is warranted. Some dilated but unobstructed infant kidneys spontaneously return to a normal or near-normal appearance with time (see previous discussion).
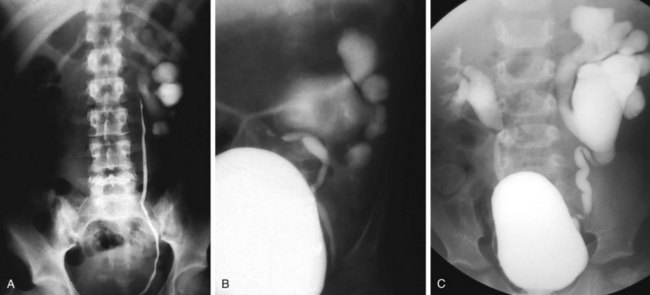
Megaureter
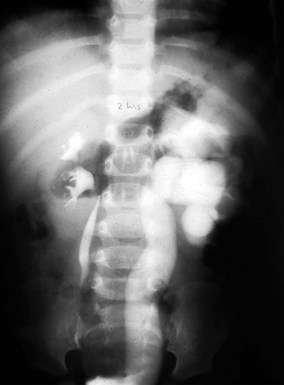
The term megaureter is descriptive of a large ureter, with or without intrarenal hydronephrosis (Fig. 14-12). Megaureters may be the result of massive vesicoureteric reflux or obstruction at the ureterovesical junction, or they may be nonobstructive. Experience with neonatal megaureters has shown that many of these lesions, when studied by diuresis renography or Whitaker protocols, are found to be nonobstructive. Some resolve spontaneously. The repair of true obstructive megaureters requires excision of the abnormal distal ureter and reimplantation of the tapered segment into the bladder. A nonrefluxing megaureter is thought to be due to either local neurologic or, more likely, muscular abnormalities of the distal ureter that interfere with normal peristalsis. Megaureters may be discovered on antenatal ultrasonography or present as a source of urinary tract infection. Calculi may form in them.
Multicystic Renal Dysplasia
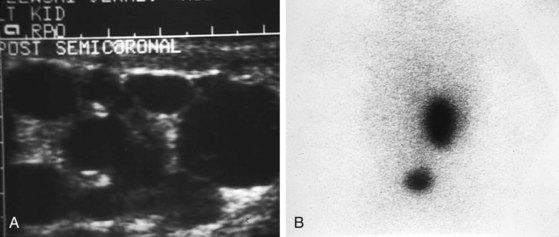
Multicystic renal dysplasia (see Chapter 13) is the second most common cause of renal enlargement in the neonate and may be discovered by antenatal ultrasound, serendipitously, or during the evaluation of an abdominal mass. Multicystic renal dysplasia must be differentiated from hydronephrosis, and the combination of ultrasonography and radionuclide scan is usually diagnostic because, although the ultrasound appearance may be similar, multicystic kidneys rarely function on scan (Fig. 14-13). Because contralateral vesicoureteric reflux is common, some authors believe that voiding cystourethrography should be performed in all patients to detect reflux into the solitary functioning kidney. A large percentage of multicystic kidneys spontaneously involute as determined by follow-up ultrasonography, and it may be that many cases of “congenitally absent kidney” represent unrecognized involution of a multicystic dysplastic kidney. There is still debate about the indications for nephrectomy in these kidneys. The Urology Section of the American Academy of Pediatrics has undertaken longitudinal follow-up of these patients. It appears that the potential for hypertension, infection, and malignancy in these kidneys is small.
Simple Renal Cysts
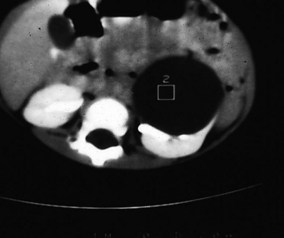
Simple renal cysts were thought to be rare in children until the advent of high-resolution ultrasound technology. They are now frequently detected, albeit much less commonly than in adults, in whom the incidence increases with age. As a result, the traditional admonition to surgically explore all cysts in children has been replaced by a policy of radiographic evaluation similar to that in adults. Simple cysts should be treated as benign. Most renal cysts are discovered serendipitously while evaluating the urinary tract for infection-related symptoms, but large cysts occasionally present as abdominal masses. Radiologic evaluation usually includes ultrasonography, but CT (Fig. 14-14) and even cyst puncture for aspiration and contrast studies may be used to confirm the nature of the cyst. The differential diagnosis includes cystic Wilms’ tumor, multilocular cystic dysplasia, duplication anomaly with hydronephrosis, caliceal diverticulum, and adult polycystic disease.
Cutaneous Urinary Diversion
Cutaneous Pyelostomy.
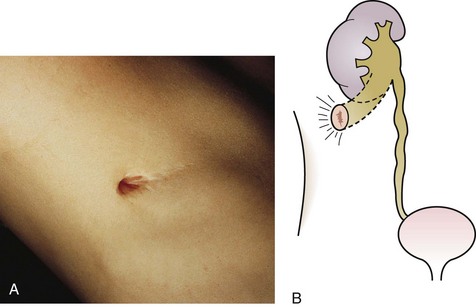
The renal pelvis is marsupialized to the skin (Fig. 14-15). This is an uncommon diversion except in small infants with severe hydronephrosis and compromised renal function.
End Ureterostomy.
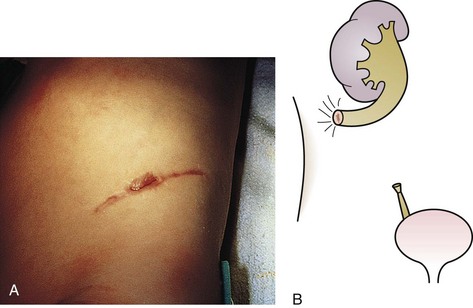
A single stoma is created, which usually requires using the distal ureter (Fig. 14-16).
Loop Ureterostomy.
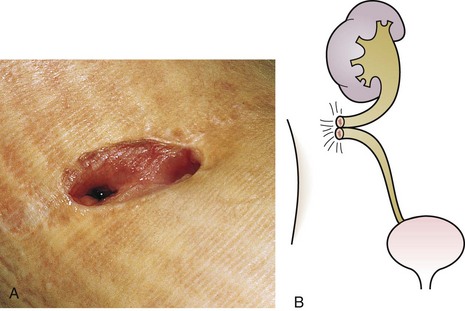
A double-barreled stoma is created, allowing access to both the proximal and distal ureter (Fig. 14-17).
Intestinal Diversion.
An isolated segment of bowel is interposed between the skin and ureters. The normal continuity of the intestinal tract is restored (Fig. 14-18).
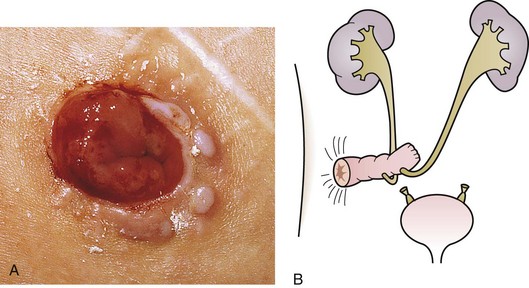
Cutaneous Vesicostomy.
This is probably the most commonly created temporary diversion in children, usually in cases of urethral valves, neuropathic bladder, prune-belly syndrome, and occasionally severe vesicoureteric reflux. A vesicostomy is basically a vesicocutaneous fistula, and in the small infant it is simply covered with a diaper (Fig. 14-19).
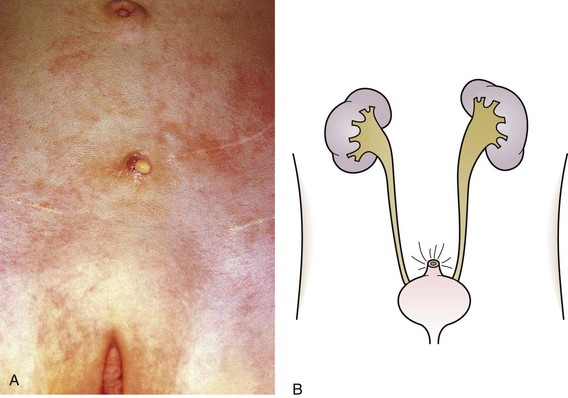
Appendicovesicostomy.
This is a continent diversion intended to allow intermittent catheterization of the bladder when urethral access is difficult (Fig. 14-20). The stoma is frequently concealed in the umbilicus.
< div class='tao-gold-member'>
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


