5 Cardiology
Physical Diagnosis of Congenital Heart Disease
Cyanosis and Clubbing
Even before mild desaturation is detectable, early clubbing and cyanosis may be seen (Figs. 5-1 and 5-2). The base of the nail, especially the thumbnail, may show loss of the angle as early as 3 months of age (see Chapter 16). Elevated hemoglobin and hematocrit and loss of nail angle indicate hypoxemia and the presence of a right-to-left intracardiac shunt (Fig. 5-3). If heart disease is excluded, chronic pulmonary disease should be considered as another potential cause of clubbing.
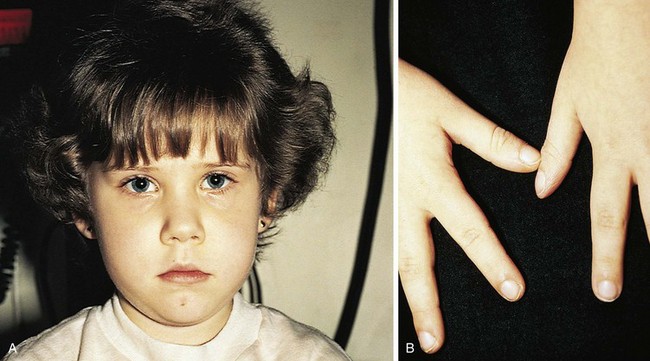
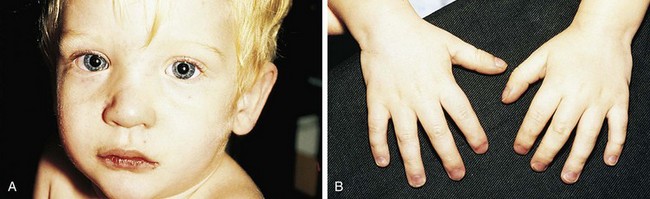
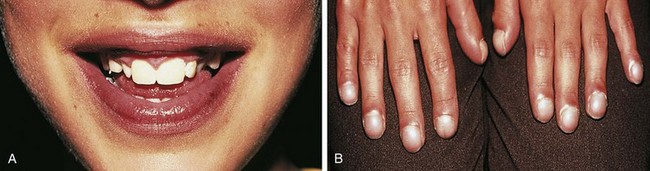
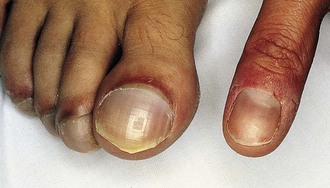
Observation of the lips and mucous membranes for the presence of cyanosis is best done in good daylight because fluorescent lighting may produce a false cyanotic tinge. In the presence of polycythemia with hemoglobin in the 18 to 20 gm/dL range and hematocrit greater than 60%, the conjunctival vessels become engorged and plethoric (see Fig. 5-2). Conversely, when a patient is anemic, visible cyanosis can be easily missed (this is particularly important in infancy, when babies reach the physiologic nadir in hematocrit). Differential cyanosis between the upper and lower extremities is an unusual clinical finding. If the patient has pulmonary vascular disease, reverse flow through a patent ductus arteriosus with no right-to-left intracardiac shunting, cyanosis, and clubbing may be found in the lower extremities but not in the hands (Fig. 5-4).
Heart Murmur Evaluation
Preschoolers and school-age children are commonly referred for evaluation of a heart murmur. Innocent murmurs of childhood fall into four major categories: systolic ejection murmurs at the base; vibratory, or Still’s, murmur; venous hums; and carotid and cranial bruits. In most instances there are associated clinical and laboratory studies that can distinguish the innocent from the pathologic murmur. Table 5-1 summarizes the distinguishing features and differential diagnosis. Figure 5-5 illustrates the sites where murmurs resulting from various cardiovascular lesions are best heard.
Table 5-1 Innocent Murmurs Mimicking Congenital Heart Disease
| Innocent Heart Murmur | Structural Congenital Heart Disease |
|---|---|
| Systolic Ejection Murmur at the Base of the Heart | |
| High left sternal border | Pulmonary valve stenosis |
| Ejection click | |
| Transmission to back | |
| Atrial septal defect | |
| Parasternal lift* | |
| S2 wide split* | |
| Diastolic murmur of tricuspid flow* | |
| High right sternal border | Aortic valve stenosis |
| Ejection click* | |
| Radiation to neck* | |
| Still Murmur | |
| Vibratory quality | Ventricular septal defect |
| Location: left midsternal border | Character of murmur* Discrete subaortic stenosis Radiation to aortic area* Subpulmonic stenosis Radiation to pulmonic area* Soft P2* |
| Venous Hum | |
| Continuous | Patent ductus arteriosus |
| Location: neck and under clavicles | Location under left clavicle* |
| Usually loudest when sitting, disappears in supine posture | No change with position* Coronary AV malformation Accentuated in diastole* |
| Carotid and Cranial Bruits | |
| Murmur over carotids and head | Aortic stenosis AV malformation Continuous murmur would support AV malformation* |
AV, atrioventricular; P2, pulmonic second sound; S2, second heart sound.
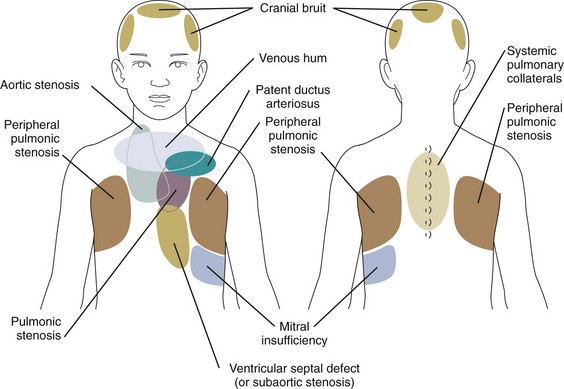
Syndrome-associated Physical Findings
Dysmorphology of the face and habitus suggests certain syndromes associated with congenital heart disease (Table 5-2).
Table 5-2 Dysmorphology Syndromes and Trisomies with Associated Cardiovascular Abnormalities
| Syndrome | Common Cardiac Defect |
|---|---|
| DiGeorge/velocardiofacial (22q11 deletion) | Aortic arch abnormalities: interrupted arch (type B), right aortic arch Conotruncal abnormalities: truncus arteriosus, tetralogy of Fallot, pulmonary atresia with ventricular septal defect |
| Ellis–van Creveld | Atrial septal defect or single atrium |
| Fetal alcohol | Ventricular septal defect |
| Holt-Oram | Atrial and ventricular septal defects, arrhythmias |
| Marfan | Dilation of ascending aorta/aortic sinus, aortic and mitral insufficiency |
| Noonan | Dysplastic pulmonic valve, atrial septal defect |
| Turner | Coarctation of the aorta, bicuspid aortic valve |
| Williams | Supravalvular aortic stenosis, pulmonary artery stenosis |
| Trisomy | |
| 13 | Patent ductus arteriosus, septal defects, pulmonic and aortic stenosis (atresia) |
| 18 | Ventricular septal defect, polyvalvular disease, coronary abnormalities |
| 21 (Down) | Atrioventricular septal defects, ventricular septal defect, patent ductus arteriosus, anomalous subclavian artery |
The typical features in Down syndrome (trisomy 21) are discussed in Chapter 1. About 40% of children with this syndrome have structural lesions such as atrioventricular septal defects (complete or partial AVSD), isolated ventricular septal defects, patent ductus arteriosus, or anomalous origin of the subclavian arteries. Although many infants with Down syndrome and congenital heart disease have chronic congestive heart failure and growth failure, there is a subset of infants with a significant septal defect who may grow and develop appropriately. This is related to abnormally high pulmonary vascular resistance not decreasing in the usual fashion in this group, leading to early pulmonary vascular disease. Because this presentation may be silent, it is important that all children with Down syndrome be thoroughly evaluated during early infancy. The evaluation should include an echocardiogram to rule out congenital heart disease.
DiGeorge and velocardiofacial syndromes are related developmental disorders involving the third and fourth pharyngeal pouches. They have been shown to be caused by deletions within a critical region of chromosome 22q11 (which can be evaluated by a blood test, namely, fluorescence in situ hybridization [FISH], for 22q11 deletion). Cardiac abnormalities, particularly the conotruncal type and aortic arch anomalies, occur in 75% of patients with 22q11 deletion (see Table 5-2). Other findings include hypocalcemia, cleft palate, renal anomalies, immunologic defects, facial dysmorphisms, and variable developmental delay with educational and behavioral issues (see Chapter 4).
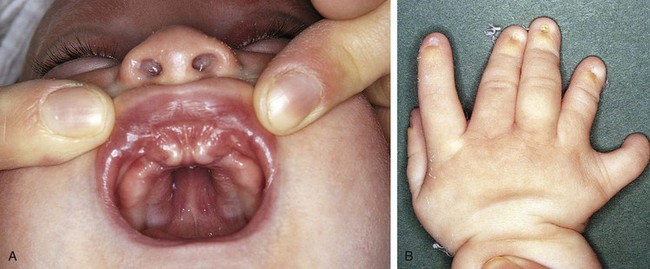
Ellis-van Creveld syndrome is an autosomal recessive disorder characterized by multiple gingival frenula, natal teeth, and polydactyly (Fig. 5-6). The patient with this syndrome frequently has an atrial septal defect or a common atrium.
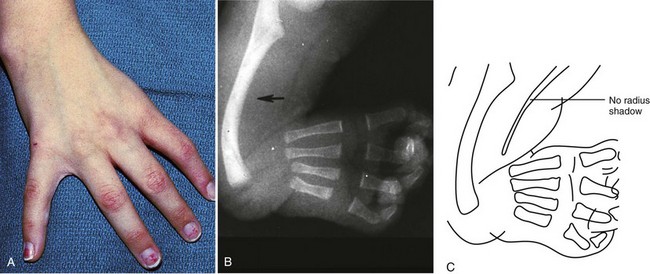
Holt-Oram syndrome, an autosomal dominant disorder, is associated with upper limb deformities consisting of narrow shoulders, hypoplasia of the radius, and phocomelia (Fig. 5-7). Absence of both radius and thumb or proximal displacement of the thumb is the most frequent finding. Commonly associated cardiovascular abnormalities include an atrial septal defect, ventricular septal defect, and arrhythmias (atrial and ventricular ectopy and atrioventricular block).
Marfan syndrome also has autosomal dominant inheritance; it manifests as a connective tissue disorder in which the elastic fibers are disrupted, causing cystic medial necrosis of the aorta, as well as joint laxity and subluxation of the ocular lens. Affected patients are tall, with increased limb length compared with the trunk. Their arm span exceeds their height. The cardiovascular abnormalities nearly always found in this syndrome include aneurysmal dilation of the aorta and aortic sinuses and mitral valve prolapse. Associated aortic and mitral valve regurgitation are common (Fig. 5-8) (see Chapter 1).
The most common cardiac defects in Turner syndrome are coarctation of the aorta and a bicuspid aortic valve. (See Chapters 1 and 9 for a detailed discussion of Turner syndrome).
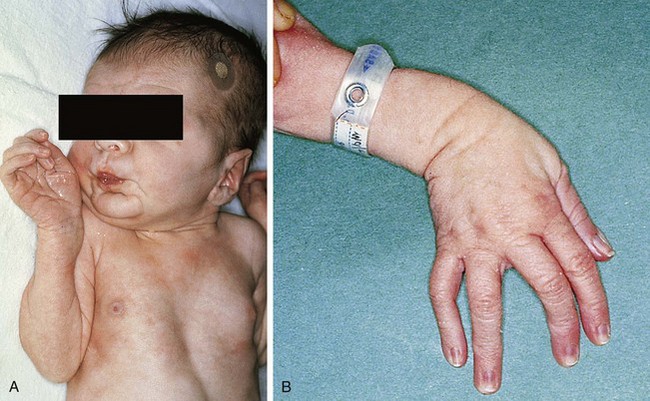
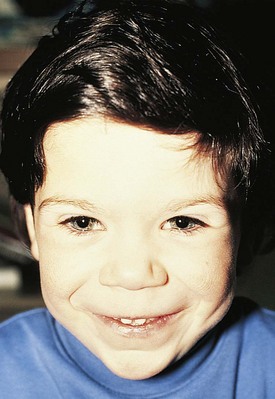
Patients with Noonan syndrome have features characteristic of Turner syndrome but possess a defect on chromosome 12 and may be male or female. Clinically, these children have the findings of webbing of the neck, pectus excavatum, shield chest with widely spaced nipples, short stature, epicanthal folds, low-set ears, and increased carrying angle of the arms (Fig. 5-9). Common cardiovascular defects include pulmonary stenosis in association with a dysplastic pulmonary valve, atrial septal defect, and hypertrophic cardiomyopathy. On occasion, there may be dysplasia of all cardiac valves. The syndrome appears as an autosomal dominant disorder; multiple members of a family are often affected.
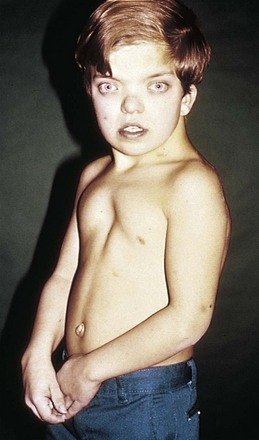
Patients with Williams syndrome characteristically have “elfin” facies: a broad maxilla, a small mandible with full mouth and large upper lip (philtrum), upturned nose, and a full forehead (Fig. 5-10). This syndrome has been associated with hypercalcemia in infants, a strikingly affable personality despite variable degrees of developmental delay, and has an identifiable genetic abnormality. Supravalvular aortic stenosis and pulmonary artery branch stenosis are the common cardiovascular abnormalities associated with this syndrome.
In addition, there are many other genetically determined diseases and inborn errors of metabolism with cardiac involvement, the most common of which are listed in Table 5-3.
Table 5-3 Other Genetic Syndromes and Inborn Errors of Metabolism with Associated Cardiovascular Findings
| Genetically Determined Diseases | Cardiac Findings |
|---|---|
| Metabolic | |
| Pompe disease (glycogen storage) | Cardiomyopathy (storage of glycogen in myocardium) |
| MPS | Storage of MPS in arteries and coronary arteries, valves with insufficiency and stenosis |
| Hurler syndrome (MPS IH), Hunter syndrome (MPS II), Scheie syndrome (MPS IS), Hurler-Scheie syndrome (MPS IH/S), Morquio syndrome (MPS IV) | |
| Hyperlipoproteinemia, familial type II | Premature atherosclerosis of arteries including coronary arteries |
| Neurologic | |
| Friedreich ataxia | Cardiomyopathy (congestive or hypertrophic) |
| Muscular dystrophies | Myocardial degeneration and fibrosis |
| Inborn Error of Metabolism (No Proven Genetic Basis) | Cardiac Findings |
| Progeria | Hypercholesterolemia, atherosclerotic changes in arteries including coronary arteries |
MPS, mucopolysaccharidosis.
Visible Clues in Acute Rheumatic Fever
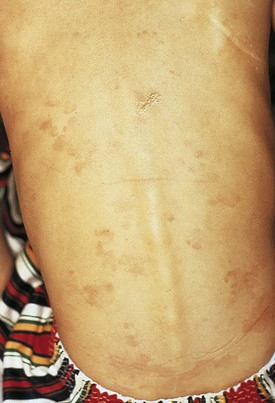
Examination of the skin in a patient with acute rheumatic fever may reveal the typical rash of erythema marginatum, although this rash is not specific for rheumatic fever. It is evanescent, nonpruritic, has sharp serpiginous margins, and is found on the inner aspects of the upper arms and thighs and on the trunk (Fig. 5-11). The differential diagnosis includes (1) drug rash, which is papular and pruritic; (2) erythema multiforme, which has target lesions; (3) rash of juvenile rheumatoid arthritis, which is pink, macular, and lacks wavy margins, and which may be transient; and (4) the cutaneous findings of Kawasaki disease (see Chapter 7).
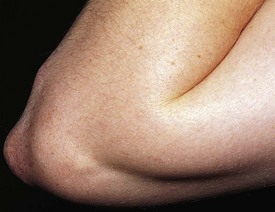
Subcutaneous nodules are rare in chronic rheumatic heart disease, but if found, they are almost always associated with severe carditis. These movable, nontender, cartilage-like swellings vary in size from 2 mm to 1 cm and are persistent. They are seen over the bony prominences of the large joints and external surfaces of the elbows and knuckles of the hands, knees, and ankles. They may also be felt along the spine and over the skull. Although difficult to photograph, they are easily palpated (Fig. 5-12).
Signs of Bacterial Endocarditis
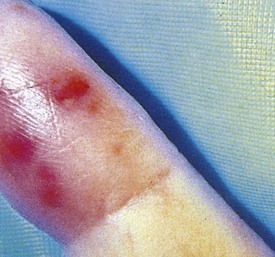
Although the clinical presentation of bacterial endocarditis varies according to the infecting organism, it should be suspected in any patient with congenital or acquired heart disease who has prolonged fever without apparent cause. The classic skin lesions include petechiae, splinter hemorrhages of the nails, conjunctival hemorrhages, and Janeway lesions (Fig. 5-13), all of which are manifestations of vasculitis. Vegetations occasionally dislodge and embolize in an end artery, which results in hemorrhagic or gangrenous lesions (Fig. 5-14). Osler’s nodes, which present as small tender erythematous nodules, are found in the intradermal pads of the fingers and toes or in the thenar or hypothenar eminences (Fig. 5-15). All the aforementioned findings are often associated with a new heart murmur, splenomegaly, spiking fever, and positive blood culture. Clubbing of the fingers may occur in chronic cases.
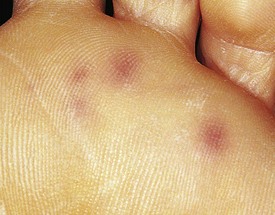
Figure 5-13 Janeway lesions. Note the small (painless) nodules on the sole of a patient with bacterial endocarditis.
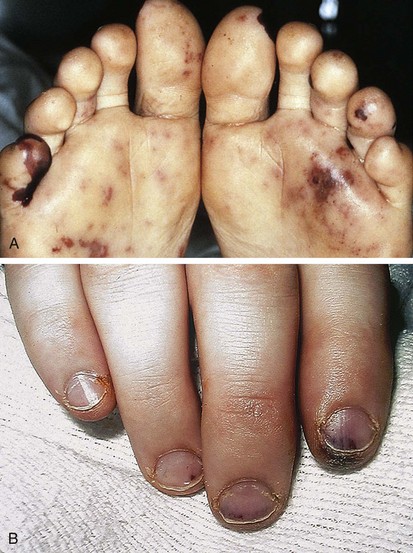
Figure 5-14 Acute bacterial endocarditis. Note the hemorrhagic lesions (A) and subungual splinter hemorrhages (B).
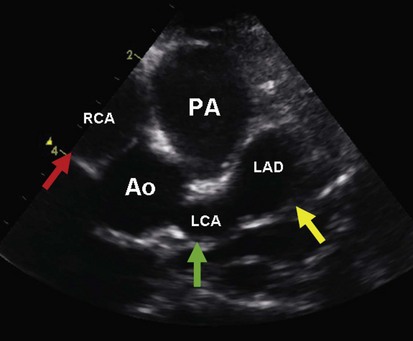
Kawasaki Disease
This multisystem disease has surpassed acute rheumatic fever as the most common form of acquired heart disease in children. It is characterized by fever, conjunctivitis, erythema of the lips and oral mucosa, extremity changes, rash, and cervical adenopathy. Coronary artery aneurysms develop in 15% to 25% of untreated patients and may result in myocardial ischemia, infarction, or sudden death. The incidence of coronary abnormalities is markedly decreased with timely treatment (~5%). This disease is discussed in detail in Chapter 7. Coronary artery aneurysms are well demonstrated by echocardiography (Fig. 5-16), and this imaging modality plays an essential role in the diagnosis and management of Kawasaki disease.
Laboratory Aids in the Diagnosis of Congenital Heart Disease
Chest Roentgenography
Cardiac Apex and Visceral Situs
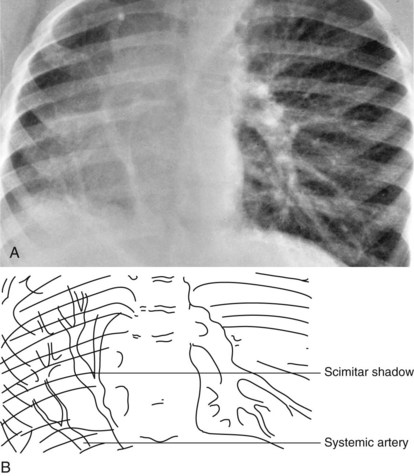
The location of the cardiac apex and visceral situs provides important diagnostic information. Discordance of the situs and cardiac apex (i.e., apex to the right with situs solitus [normal arrangement] or apex to the left with situs inversus) is often associated with structural congenital heart disease (Figs. 5-17 and 5-18). Dextrocardia (apex to the right) or mesocardia (apex to the middle) with situs solitus is a frequent presentation of ventricular inversion or corrected transposition of the great arteries (see Fig. 5-17). Dextrocardia can also be seen with primary pulmonary problems. Scimitar syndrome is composed of dextrocardia with hypoplasia of the right lung (Fig. 5-19). In this case a major portion of the right lung (usually the right lower lobe) is “sequestered” and has its arterial supply by way of a systemic artery from the descending aorta, and the pulmonary venous return from that lung drains abnormally into the inferior vena cava via a vein forming a scimitar (see Fig. 5-19). Patients with levocardia (apex to the left) with either situs inversus or situs ambiguus frequently have complex congenital heart diseases such as transposition of the great arteries, pulmonary atresia, and atrioventricular septal defects. Atrial isomerism is associated with bilateral morphologic right or left lungs and can be recognized as bilateral symmetrical right (short) or left (long) bronchi. This is best demonstrated with a magnified penetrated chest x-ray examination focusing on bronchial anatomy (Fig. 5-20). Almost all patients with this anomaly have complex congenital heart disease.
Shape and Size
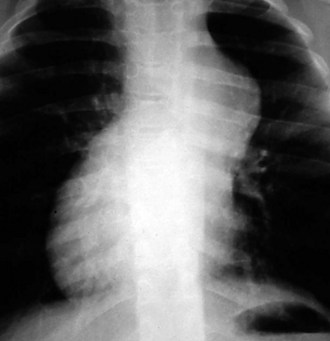
Cardiac size is important, but the shape of the cardiac image may also provide a clue as to which heart chambers are enlarged and the likely structural diagnosis. In the cyanotic newborn with transposition of the great arteries, the cardiac image appears as an “egg on a string” (Fig. 5-21). If the thymic shadow does not obscure it, the mediastinal shadow shows a narrow waist resulting from the posteromedial position of the main pulmonary artery. This produces the “string.” Pulmonary vascular markings are usually increased, although vascularity may be normal in the immediate newborn period.
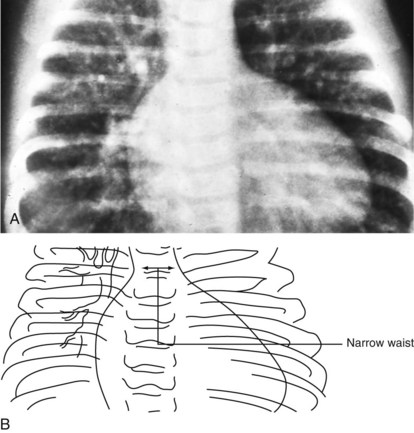
In tetralogy of Fallot with pulmonic stenosis, the heart appears “boot-shaped” because right ventricular hypertrophy causes the apex (toe of the boot) to turn upward (Fig. 5-22). The concavity of the left upper cardiac border is due to the small right ventricular outflow tract and main pulmonary artery segment.
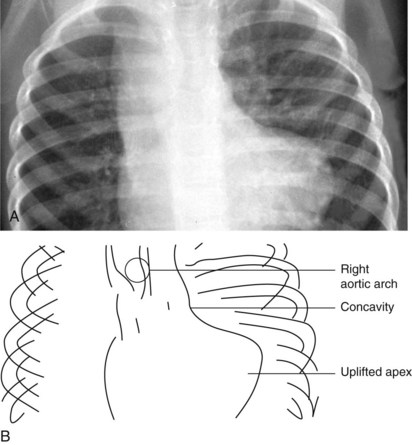
In tetralogy of Fallot with pulmonary atresia, the heart is shaped like an “egg on its side” (Fig. 5-23). The pulmonary blood flow to the lungs may be supplied by either a patent ductus arteriosus or systemic arterial collateral vessels. The pulmonary vascular markings are decreased if pulmonary blood flow is patent ductus dependent and increased if large systemic collaterals supply pulmonary blood flow.
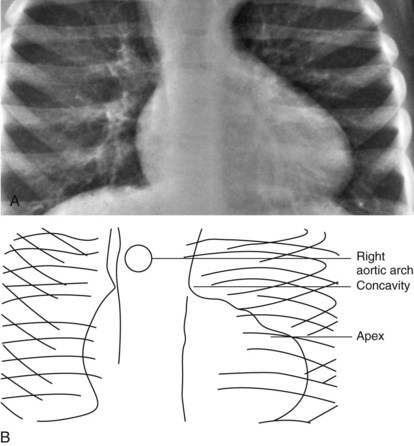
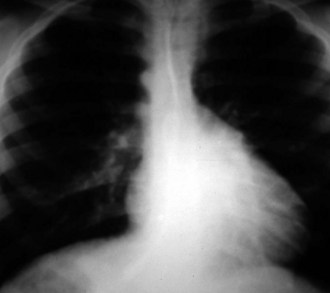
In congenitally corrected transposition of the great arteries, the heart has a “valentine” or “heart” shape with the apex pointing downward just to the left of the midline, as shown in Figure 5-24. The fullness at the left upper border of the cardiac shadow is due to the ascending aorta arising from the left-sided morphologic right ventricle.
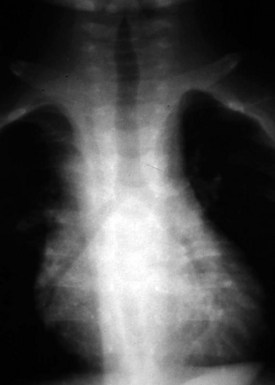
Although an enlarged and globular heart shadow may be associated with a cardiomyopathy, massive cardiac enlargement (so-called “wall-to-wall” heart) is typical in patients with Ebstein anomaly. In this anomaly there is a malformation of the tricuspid valve with downward displacement of the inferior and septal leaflets of the valve into the ventricle, causing severe tricuspid valve regurgitation or stenosis. As a result, the right atrium becomes markedly enlarged and, along with the “atrialized” portion of the right ventricle, contributes significantly to the cardiac image of a large box-shaped heart (Fig. 5-25).
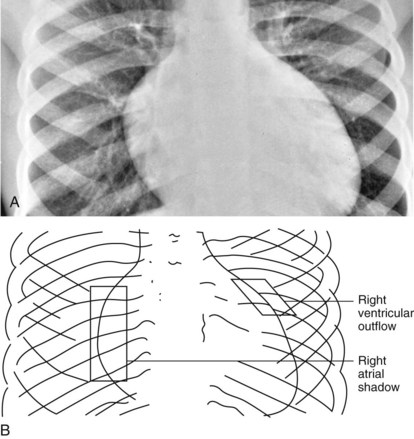
Left-to-right shunt lesions from atrial septal defects, ventricular septal defects, or a patent ductus arteriosus demonstrate specific chamber enlargement and increased pulmonary vascular markings. A significant atrial defect shows enlargement of all right-sided cardiac chambers including the right atrium, right ventricle, and pulmonary artery (Fig. 5-26). A patent ductus arteriosus shows enlargement of all left-sided cardiac chambers including the aorta. In patients with a ventricular septal defect, the right atrium is the only heart chamber that is not enlarged.
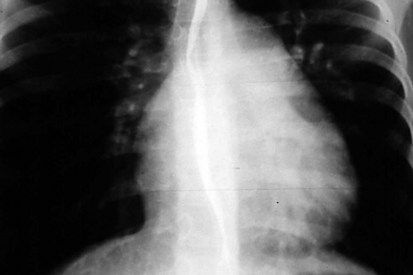
Great Vessels
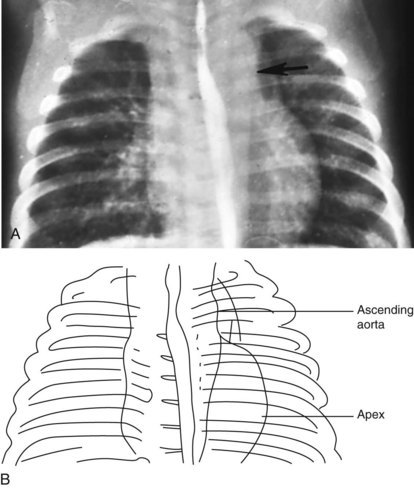
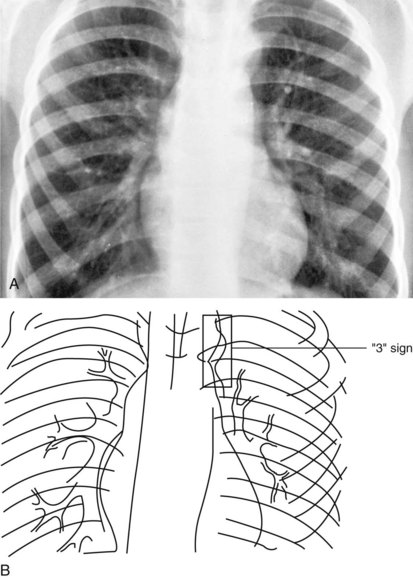
The radiographic appearance of the great arteries may also suggest a specific structural congenital heart defect. The main and left branch pulmonary arteries are usually enlarged in patients with pulmonary valve stenosis, due to poststenotic dilation (Fig. 5-27). The characteristic radiographic finding of congenital aortic valve stenosis is dilation of the ascending aorta, best seen as an overlapping shadow with the superior vena cava along the right upper cardiac border (Fig. 5-28). Coarctation of the aorta not diagnosed in a timely fashion may show the distinct radiographic finding of a “reversed E” or “3” sign caused by prestenotic and poststenotic dilation of the descending aorta (Fig. 5-29). If a significant coarctation remains unrepaired for 5 to 10 years, rib notching may appear (see the section Skeletal Abnormalities, below).
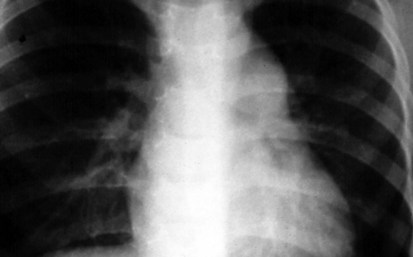
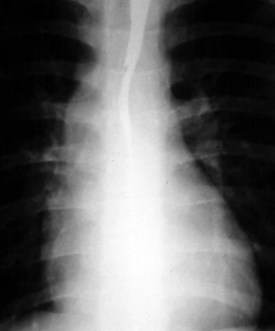
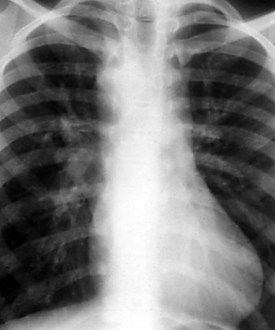
The normal left aortic arch causes a shift of the tracheal air column to the right, whereas a right arch causes a similar deviation to the left (Fig. 5-30). The position of the thoracic descending aorta also helps define the side of the arch and can be determined by noting obscuring of either the left or right side of the thoracic vertebral bodies. A right aortic arch should always raise the suspicion of congenital heart disease and is found in approximately 30% of patients with tetralogy of Fallot or truncus arteriosus.
< div class='tao-gold-member'>
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


