Chapter 337 Tumors of the Digestive Tract
Tumors of the digestive tract are mostly polypoid. They are also commonly syndromic tumors and tumors with known genetic identification (see  Table 337-1 on the Nelson Textbook of Pediatrics website at www.expertconsult.com). They usually manifest as painless rectal bleeding, but they can serve as lead points for intussusception.
Table 337-1 on the Nelson Textbook of Pediatrics website at www.expertconsult.com). They usually manifest as painless rectal bleeding, but they can serve as lead points for intussusception.
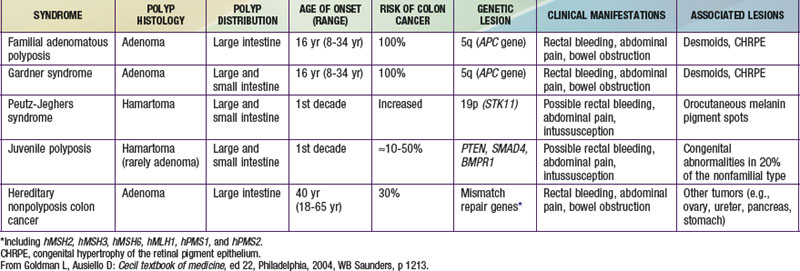
Peutz-Jeghers Syndrome
Peutz-Jeghers syndrome (PJS) is a rare autosomal dominant disorder (incidence, ∼1 : 120,000) characterized by mucocutaneous pigmentation and extensive GI hamartomatous polyposis. Macular pigmented lesions may be dark brown to dark blue and are found primarily around the lips and oral mucosa, although these lesions may also be found on the hands, feet, or perineum (Fig. 337-1). Lesions can fade by puberty or adulthood.
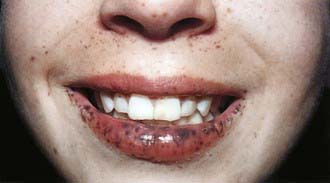
Figure 337-1 Peutz-Jeghers syndrome. Note the pigmentary changes.
(From Swartz MH: Textbook of physical diagnosis: history and examination, ed 4. Philadelphia, 2002, WB Saunders, p 295.)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


