Fig. 1
The spinal cord is comprised of ventral and dorsal white and grey matter. The ventral grey matter of the spinal cord contains the alpha motor neuron. (Reproduced with kind permission from Marion Murray and Springer Science + Business: Neuroscience in Medicine, chapter “Ulnar Deficiencies,” 2008, page 209, Murray M, Figure 13)
Basic Peripheral Nerve and Brachial Plexus Anatomy
Once the ventral and dorsal nerve roots exit the spinal cord, they form the spinal nerve roots, which join in the periphery to form the plexus or continue on as peripheral nerves. The majority of NA patients present with brachial plexus involvement. However, the lumbosacral plexus, phrenic nerve, recurrent laryngeal nerve, cranial nerves, and the distal autonomic nervous system may be involved, especially in HNA. The brachial plexus, Fig. 2, formed from the cervical spinal nerve roots C5, C6, C7, C8, and T1, provides the sensory and motor innervation for the upper extremity. It is located between the neck and axilla, proximally between the anterior and middle scalene muscles and distally just posterior to the clavicle and pectoralis muscles.
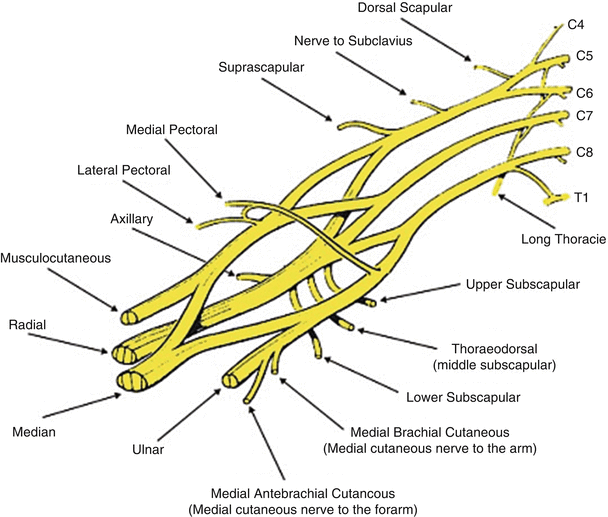
Fig. 2
Cervical and thoracic nerve roots form the brachial plexus by branching into the upper, middle, and lower trunks, then on to the anterior and posterior divisions, which further merge to become the lateral, medial, and posterior cords. The cords then branch into the peripheral nerve distributions, the musculocutaneous, axillary, radial, median, and ulnar nerves (Reproduced with kind permission from Marios Loukas and Springer Science + Business Media: Surgical and Radiologic Anatomy, The prefixed and postfixed brachial plexus : a review with surgical implications, volume 32, issue 3, 2010, page 253, Pellerin M, Kimball Z, Tubbs RS, Nguyen S, Matusz P, Cohen-Gadol AA, Loukas M)
The plexus is divided into roots, trunks, divisions, cords, and branches moving distally. The nerve roots C5, C6, C7, C8, T1, and variably C4 and T2 descend from the spinal cord through the neural foramen into the neck to form the plexus. C5 and C6 form the upper trunk, C7 the middle trunk, and C8 and T1 the lower trunk. The trunks descend and each branches into an anterior and a posterior division. The anterior divisions of the upper and middle trunks join to form the lateral cord. The posterior divisions from the upper, middle, and lower trunk join to form the posterior cord. The anterior division from the lower trunk continues on to form the medial cord. Off of the lateral cord branch the lateral pectoral nerve and the musculocutaneous nerve. It also joins the medial cord to form the median nerve. The posterior cord continues on to become the radial and axillary nerves. The medial cord continues on to become the ulnar nerve (Preston and Shapiro 2005).
On a more cellular level, these peripheral nerves are made up of a layer of connective tissue immediately surrounding the myelin sheath of an axon, called the endoneurium. The endoneurium-covered nerves are further grouped together and covered by another layer of connective tissue, the perineurium. The perineurium-covered nerves are then grouped together and covered by a final layer of connective tissue, the epineurium, to form a nerve.
Inflammatory and Autoimmune Etiology
The pathophysiology of NA, though not completely understood, is thought to be inflammatory and autoimmune in nature. There may also be a mechanical component related to local repetitive trauma.
On a cellular level, biopsies of affected brachial plexi demonstrate mononuclear inflammatory infiltrates, mainly T-lymphocytes, surrounding the epineurial and endoneurial vessels (Suarez et al. 1996). This inflammatory infiltration causes patchy damage to the brachial plexus and other nerves leading to the characteristic presentation of patchy and severe neuropathic pain followed by muscle paralysis.
It is often preceded by a viral or bacterial infection, such as an upper respiratory infection. A history of immunization, serum therapy, pregnancy, childbirth, or surgery may also antecede symptoms. With any of the aforementioned, the body may produce an inappropriate immune-mediated response against the brachial plexus resulting in nerve inflammation and subsequent injury (van Alfen et al. 2000a).
In addition to inflammatory and autoimmune pathophysiology, both INA and HNA can be related to local trauma to the neck involving the brachial plexus. On a more cellular level, local trauma can weaken the perineurium resulting in focal damage to the individual fascicles of the nerve subsequently resulting in the scattered pattern of motor and sensory involvement (van Alfen 2011).
Similarly, idiopathic TM is thought to be autoimmune in nature with multiple theories of pathophysiology, all of which culminate in the demyelination and neuronal injury of the spinal cord. Infiltrates in the spinal cord in patients with TM include monocytes and CD4+ and CD8+ lymphocytes, which are thought to lead to necrosis and cavitation of the spinal cord. The theory of molecular mimicry is evidenced in the often preceding infection (Lyme’s disease, HIV, mycoplasma, herpes virus, syphilis, and other central nervous system infections) leading to anti-GM1 antibodies and the subsequent attack by the body’s own immune system. The theory of superantigens is that certain infections induce T-cells against myelin causing destruction of the spinal cord. There may also be humoral-mediated dysregulation leading to increased IgE causing an allergic response and further tissue destruction. Finally, IL-6 released from astrocytes and microglia bind to oligodendroglia and axons causing activation of nitric oxide synthetase leading to tissue damage of the spinal cord (Wolf et al. 2012).
In addition to the aforementioned, disease-associated myelitis is found in conjunction with a number of autoimmune diseases such as sarcoidosis, Behçet’s disease, Sjögren’s syndrome, connective tissue disorders, and systemic lupus erythematosus. There is also a spectrum of inflammatory demyelinating disorders of which TM is a part of, in addition to NMO, MS, and ADEM. NMO is defined as transverse myelitis found in conjunction with optic neuropathy. NMO is a well-known autoimmune disorder and is associated with AQP4, aquaporin, antibodies. Acute myelitis may also be the presenting symptom in MS, and thus children may be initially diagnosed with TM only to further go on to be diagnosed with MS upon further imaging or another demyelinating episode. Similar and yet distinct, there is now a recurrent form of acute TM, making this distinction sometimes difficult (Borchers and Gershwin 2012).
Assessment
Signs and Symptoms
The majority of children with NA present after a viral upper airway infection, osteomyelitis of the shoulder or humerus (most commonly seen in neonates), or vaccination. Two thirds of children present with severe neuropathic pain in the shoulder or arm, which is consistent with adult presentation. The other third present with painless weakness and atrophy of the shoulder girdle or, less often, more distal muscles of the upper extremity (van Alfen et al. 2000a). If present, the initial pain may last approximately 2–3 weeks followed by patchy paralysis and atrophy of the muscles innervated by the affected nerves (Tsairis et al. 1972). One should consider other etiologies such as tumor, which can infiltrate local lymph nodes resulting in compression of the brachial plexus, direct infiltration of the nerve by lymphoma or leukemia, as well as primary nerve sheath tumors such as schwannomas, neurofibromas, or neurofibrosarcomas. These usually result in more insidious onset of pain, paresthesias, and muscle atrophy, but should be considered in the differential diagnosis.
TM is also often preceded by infection but less commonly following vaccination (Wolf et al. 2012). The most frequent initial symptoms are fever; pain in the back, extremities, and abdomen; ascending numbness and weakness; walking difficulty; balance problems; and loss of bowel and bladder control (urinary retention and/or constipation). The distribution of these symptoms is dependent on the spinal level of the lesion as well as the area of the spinal cord involved. Cervical cord lesions present with upper extremity as well as lower extremity weakness and sensory deficits; lower thoracic and lumbar lesions result in lower extremity weakness and numbness only. Initial sensory loss and weakness are ascending and may present similarly to Guillain-Barré syndrome. If the posterior columns are involved, the child may have fine motor dyscoordination leading to ataxia (Borchers and Gershwin 2012; Wolf et al. 2012). In the subgroup of children with alpha motor neuron involvement or root level involvement, the typical signs and symptoms may be present initially, but are followed by recovery of all except one limb, which remains flaccid, atrophic, and areflexic (Sadowsky et al. 2011).
In the initial phase of TM, patients often present in spinal shock with absent deep tendon reflexes (DTRs) and interruption of the sympathetic flow through the spinal cord resulting in bradycardia and hypotension. Spinal shock generally resolves in a period of days to 12 weeks with return of DTRs and signs consistent with UMN findings including hyperreflexia, Babinski’s reflex, and increase in muscle tone.
With lesions above T6, there is a risk of autonomic dysreflexia. Other signs and symptoms involve urinary retention secondary to disruption of the connection between the pontine micturition center and the sacral spinal cord. Constipation is also a potential complication of neurogenic bowel secondary to decreased motility.
Physical examination in a patient suspected to have NA includes a complete generalized examination including overall appearance as well as evaluation for hypotelorism or other facial dysmorphic features, which are associated with HNA (Jeannet et al. 2001).
Head, eyes, ears, nose, and throat evaluation must be completed to look for signs of viral or bacterial infection, which may precede the onset of pain and weakness in NA and TM. In NA, evaluate speech for possible dysphonia with recurrent laryngeal nerve involvement. Palpation of the neck to evaluate for mass lesions of the brachial plexus is pertinent as well. This should be followed by a complete cardiovascular and respiratory examination, as involvement of the phrenic nerve may result in diaphragm paralysis and result in paradoxical breathing patterns. Also look for vasomotor symptoms such as edema, nail and hair changes, and temperature dysregulation (Fig. 3).
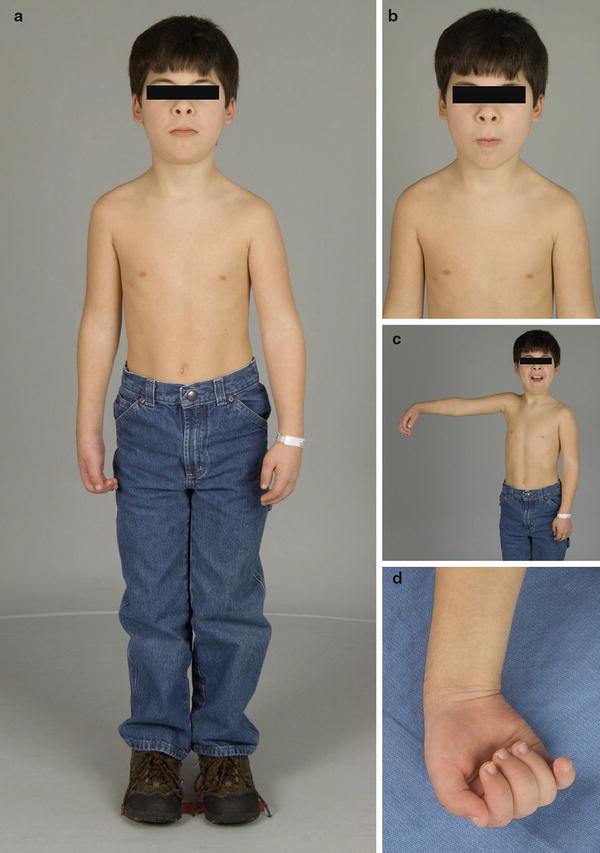
Fig. 3
Ten-year-old male with recurrent hereditary neuralgic amyotrophy (HNA) (Courtesy of Shriners Hospital for Children, Philadelphia). (a) Episode with right-sided weakness. (b) Typical facial features with considerable hypotelorism. (c) Decreased shoulder abduction and wrist drop. (d) Limited ability to grasp
General examination should be followed by a full neurologic and musculoskeletal examination. Start the neurologic examination by testing the function of the cranial nerves I-XII as NA may affect these. One should then test sensation to light touch and pinprick in both a dermatomal fashion to rule out cervical cord or root pathology, but also in the peripheral nerve distribution to evaluate for plexus or peripheral nerve injury. DTRs and other primitive reflexes should be assessed to evaluate for hyperreflexia or hyporeflexia to determine if there is UMN or LMN involvement. Peripheral nerve lesions resulting from NA should only be associated with lower motor neuron findings. In TM, the majority of cases may have UMN signs, though some cases may present with alpha motor neuron involvement resulting in LMN exam findings (Fig. 4).
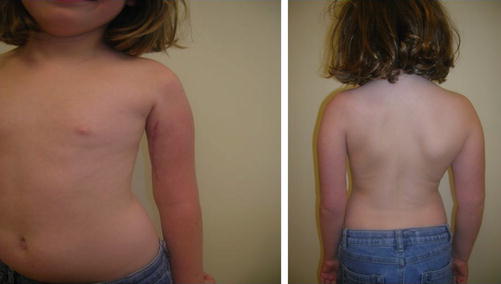
Fig. 4
A 3-year-old child with residual flaccid monoplegia of the left upper extremity related to transverse myelitis involving the alpha motor neuron of the ventral spinal cord. Muscle atrophy and circumferential and length differences can be present. This is postsurgical intervention, as evidenced by the surgical scar on the anteromedial arm (Reproduced with permission from Sadowsky et al. (2011))
Manual muscle testing should be completed and classified. One may use the Hospital for Sick Children Active Movement Scale (Tables 1 and 2). Scoring ranges from 0 to 5 with 0 = no muscle contraction with gravity removed, 1 = flicker of movement with gravity removed, 2 = less than 50 % range of motion (ROM) with gravity removed, 3 = greater than or equal to 50 % ROM with gravity removed, 4 = full ROM with gravity removed, and 5 = less than 50 % ROM against gravity.
Table 1
The active movement scale and myotomal distribution (Leis 2010)
Action | Muscles | Myotomal distribution |
|---|---|---|
Shoulder abduction | Middle deltoid, supraspinatus | C5, C6 |
Shoulder adduction | Pectoralis major, latissimus dorsi, teres major, coracobrachialis, infraspinatus, long head of the triceps, anterior and posterior deltoid | C5, C6, C7, C8, T1 |
Shoulder flexion | Anterior deltoid, pectoralis major, biceps brachii, coracobrachialis | C5, C6, C7, C8, T1 |
Shoulder external rotation | Infraspinatus, teres minor, posterior deltoid, supraspinatous | C5, C6 |
Shoulder internal rotation | Subscapularis, pectoralis major, latissimus dorsi, anterior deltoid, teres major | C5, C6, C7, C8, T1 |
Elbow flexion | Biceps brachii, brachialis, brachioradialis, pronator teres | C5, C6, C7 |
Elbow extension | Triceps, anconeus | C6, C7, C8, T1 |
Forearm supination | Biceps brachii, supinator | C5, C6 |
Forearm pronation | Pronator quadratus, pronator teres, flexor carpi radialis | C6, C7, C8, T1 |
Wrist flexion | Flexor carpi ulnaris, flexor carpi radialis, palmaris longus, flexor digitorum superficialis, flexor digitorum profundus, flexor pollicis longus | C6, C7, C8, T1 |
Wrist extension | Extensor carpi ulnaris, extensor carpi radialis longus, extensor carpi radialis brevis, extensor digitorum communis, extensor digiti minimi, extensor indicis, extensor pollicis longus | C6, C7, C8, T1 |
Finger flexion | Flexor digitorum superficialis, flexor digitorum profundus, lumbricals, dorsal and palmar interossei, flexor digiti minimi | C7, C8, T1 |
Finger extension | Extensor digitorum communis, extensor indicis proprius, extensor digiti minimi | C7, C8 |
Thumb flexion | Flexor pollicis brevis, flexor pollicis longus, opponens pollicis, adductor pollicis | C8, T1 |
Thumb extension | Extensor pollicis longus, extensor pollicis brevis, abductor pollicis longus | C7, C8 |
Table 2
Hospital for sick children active movement scale
Muscle grade | Definition |
|---|---|
0 | No muscle contraction with gravity removed |
1 | Flicker of movement with gravity removed |
2 | Less than 50 % range of motion (ROM) with gravity removed |
3 | Greater than or equal to 50 % ROM with gravity removed |
4 | Full ROM with gravity removed |
5 | Less than 50 % ROM against gravity |
The musculoskeletal examination begins with inspection of the child for edema, ecchymosis, deformity, scar, rash, or atrophy. It is important also to evaluate muscle bulk, scapular positioning, arm positioning (abduction, adduction, internal rotation, external rotation), and the child’s posture at rest and with movement. Palpation starts with the bony structures including the sternal notch, the sternoclavicular joint, along the clavicle to the acromioclavicular joint, and on to the greater and lesser tuberosities of the humerus. From here palpate the coracoid process, the suprascapular fossa, and along the spine of the scapula. To examine the scapula, ask the child to flex both arms and push against a wall. Evaluate scapular movement; if the scapula wings medially, then the serratus anterior (innervated by the long thoracic nerve) is likely weak and/or atrophic, resulting in unopposed middle trapezius (innervated by CN XI) action (Fig. 5).
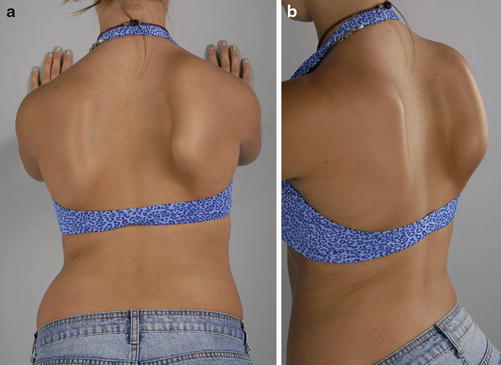
Fig. 5
Fourteen-year-old female with neuralgic amyotrophy affecting the right long thoracic nerve (Courtesy of Shriners Hospital for Children, Philadelphia). (a) Right scapular winging exacerbated by pushing against wall. (b) Side view with marked scapular winging
With abduction of the arm, a laterally deviating scapula is indicative of upper trapezius weakness and unopposed serratus anterior muscle. Special musculoskeletal tests to evaluate supraspinatus muscle impingement or bicipital tendinitis should be considered to rule out other soft tissue-related causes (Malanga and Nadler 2006). In addition to the aforementioned musculoskeletal examination, the Mallet classification, the Toronto Test Score, and the Active Movement Scales (AMS), as mentioned above, may be used to measure active movement of the upper extremity (Bae et al. 2008).
Imaging and Other Diagnostic Studies
Diagnosis of NA is made with a combination of blood work, imaging studies, and electrodiagnostic studies, though it remains a diagnosis of exclusion. Laboratory studies, though unnecessary for diagnosis, may show mildly elevated creatine kinase and elevated liver function tests. Other signs of prior infection, such as viral titers or antibodies, may be positive. As in other autoimmune disorders, antiganglioside antibodies may also be present in the serum. Cerebrospinal fluid (CSF) studies in NA are generally normal, except for slightly increased CSF protein (van Alfen 2007).
In addition to the aforementioned laboratory studies, imaging studies may also contribute to the diagnosis not only by ruling out other pathologies but also in findings consistent with NA. Imaging may begin with a basic chest X-ray to evaluate for the presence of a mass lesion or for elevation of the diaphragm as seen with phrenic nerve involvement. For further evaluation, magnetic resonance neurography (MRN) of the neck to evaluate the plexus directly and MRI of the shoulder to evaluate the musculature are appropriate. The MRN of the brachial plexus may demonstrate T2-signal enhancement or focal thickening of the plexus (van Alfen and van Engelen 2006). MRI of the neck and shoulder may show denervation of affected muscles with findings of intramuscular edema, muscular atrophy, and fatty infiltration. The most commonly affected muscles seen on MRI are the supraspinatus and the infraspinatus. Though it is not completely defined, in the acute phase of denervation, there may not be positive findings on MRI. The first sign may be diffuse increased signal related to edema seen on T2-weighted images of involved musculature (Scalf et al. 2007).
Diagnostic criteria for TM have been defined by the TMCWG as described previously and include imaging, CSF studies, and serological studies. Serum evaluation includes mycoplasma antibodies, West Nile Virus titers, Bartonella henselae titers, and Lyme titers to evaluate for infection. Rheumatological blood work should include rheumatoid factor, antinuclear, anti-double-stranded DNA, anti-single-stranded DNA, anti-RNP, anti-smooth muscle, anti-SSA, anti-SSB, and antiphospholipid antibodies to evaluate for autoimmune disorders. Aquaporin-4 antibodies or NMO IgG should be evaluated for the possibility of NMO-associated myelitis. CSF studies should demonstrate pleocytosis or increased IgG. CSF cultures should be sent to rule out for infectious myelitis (Wolf et al. 2012).
MRI of the spine demonstrates pathology in 78 % of cases of clinical TM with lesions most commonly in the cervicothoracic region of the spinal cord (Fig. 6). Some patients have multifocal lesions, though more commonly they are singular lesions. Two thirds of lesions involve the gray matter only and one third both gray and white matter. The majority of lesions span three or more segments of the spinal cord, and in the acute phase, 19.1–62.5 % enhance with gadolinium (Alper et al. 2011; Sellner et al. 2009).
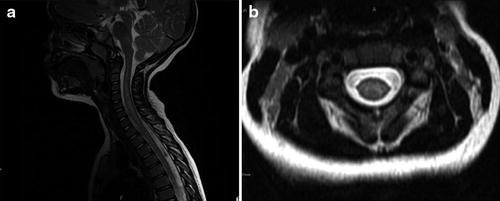
Fig. 6
T2-weighted imaging of the cervical spinal cord of child with residual flaccid monoplegia after idiopathic TM with cervical cord inflammation involving the alpha motor neuron. (a) Longitudinal involvement of the cervical spinal cord from C3 through C7. (b) Involvement of the ventral spinal cord and thus the alpha motor neuron (Reproduced with permission from Sadowsky et al. (2011))
Electrodiagnostic studies such as nerve conduction studies (NCS) and electromyography (EMG) may be useful in diagnosing NA, when performed 10–14 days after onset of symptoms to avoid false negatives. NCS may show normal to slightly prolonged conduction velocities with or without decreased amplitude of the sensory nerve action potentials (SNAP) and compound motor action potentials (CMAP). EMG may show axonal loss secondary to acute denervation with fibrillations, positive sharp waves, and polyphasic high-amplitude motor unit action potentials (MUAP). There are rare instances, however, that demonstrate a pattern of demyelination instead of axonal loss (Vassallo et al. 2010). Since TM is generally an UMN disease process, it usually would not demonstrate any findings on EMG. However, in the subset of patients with flaccid monoplegia resulting from damage to the alpha motor neuron, NCS/EMG may demonstrate a severe motor neuronopathy with acute and chronic denervation findings (Sadowsky et al. 2011).
Associated Injuries
The initial pain associated with NA infrequently persists, but a small subset of children may go on to develop a chronic pain syndrome with either neuropathic or musculoskeletal-type pain or a combination of the two. The musculoskeletal pain may be related to the atrophy and resultant compensatory mechanisms and poor body mechanics necessary for the child to function as normally as possible (van Alfen 2007). The neuropathic pain may be a continuation of the pain at initial onset. In HNA, there is a subset of patients that have a relapsing and remitting disease course with exacerbations every 6½ years resulting in pain and musculoskeletal dysfunction.
TM-associated injuries in the acute phase of the disease process may include loss of upper airway patency and dysphagia secondary to cranial nerve IX (glossopharyngeal nerve) involvement in the innervation of the pharyngeal muscles. Dysphagia may also be related to longitudinally extensive lesions with extension into the brainstem. If the spinal lesion is above C5, weakness of the diaphragm may ensue, leading to respiratory difficulty. In the longer term, if the lesion is above T6, there is a risk of autonomic dysreflexia (AD), which is a sympathetic discharge resulting from a noxious stimuli below the level of the injury (such as distended bladder or stool impaction) and resulting in hypertension, sweating, headache, goose bumps, and flushing. This is a medical emergency and is treated by removing the noxious stimulus and, if no improvement, lowering the blood pressure with pharmacological agents per published guidelines (Consortium for Spinal Cord Medicine 2011; Wolf et al. 2012).
Classification
NA is divided into two distinct categories: idiopathic and hereditary neuralgic amyotrophy. INA, the more common of the two, presents, as mentioned above, with severe pain usually in the shoulder followed by muscle paresis, atrophy, and sensory deficits. This is a singular episode that resolves over the course of months to years.
HNA, an autosomal dominant form of NA linked to a mutation in the SEPT9 gene on chromosome 17q25, is set apart by its recurring attacks of the characteristic acute severe pain. This is also followed by muscle weakness, atrophy, and sensory deficits. It predominantly involves the brachial plexus but may also affect the lumbosacral plexus, cranial nerves, phrenic nerve, recurrent laryngeal nerve, and the autonomic nervous system or a combination. Any further differences are discussed throughout the chapter.
TM is classified in a number of ways. It is classified as disease-associated transverse myelitis when found in conjunction with signs and symptoms of MS, ADEM, NMO, and rheumatologic conditions such as SLE or antiphospholipid antibody syndrome. Idiopathic TM is a diagnosis of exclusion and accounts for the majority of pediatric cases of TM (Wolf et al. 2012).
It can be further classified as acute partial TM (APTM), acute complete TM (ACTM), and longitudinally extensive TM (LETM), based on MRI findings. APTM is defined by lesions of the spinal cord on MRI that are asymmetric, patchy, and span fewer than two vertebral segments with resultant mild to moderate weakness, asymmetric sensory loss, and possible bladder involvement. These patients may go on to develop MS with multiple lesions separated over time and space throughout the central nervous system. ACTM results in moderate to severe symmetric sensory and motor deficits secondary to lesions that span the spinal cord. LETM is defined as a longitudinally extensive lesion that spans at least three vertebral segments. These patients, though at very low risk for developing MS, have a high possibility of progressing to NMO. Often the first lesion in NMO is a longitudinally extensive spinal cord lesion; thus aquaporin-4 (or NMO IgG) antibodies are important diagnostic tools in these patients. Relapse of TM may be more common in APTM compared to ACTM as well as in LETM with a diagnosis of NMO. Recurring TM is very rare. Therefore patients should have a reevaluation by an experienced neurologist to assess for possibility of additional progression or diagnosis of MS, since this may impact function and prognostication (Borchers and Gershwin 2012).
Outcome Tools
The Functional Independence Measure in Children (WeeFIM®) and the Modified Rankin Scale (MRS) are general functional measures used in children to determine the level of independence in daily life activities. The WeeFIM® measures self-care (eating, grooming, bathing and dressing), sphincter control (toileting, bowel and bladder management), transfers (chair/wheelchair, toilet, tub/shower), locomotion (walk, wheelchair, crawl, stairs), communication (comprehension, expression), and social cognition (social interaction, problem solving, memory). WeeFIM® levels range from complete independence all the way to total assistance on a seven-point scale. This is used in children with NA and TM to determine their general level of function as it pertains to their daily life (Msall et al. 1994). The MRS, developed for adults, may also be used as a general measure of function and ranges from 0 = no symptoms at all; 1 = no significant disability despite symptoms, able to carry out all usual duties and activities; 2 = slight disability, unable to carry out all previous activities, but able to look after own affairs without assistance; 3 = moderate disability, requiring some help, but able to walk without assistance; 4 = moderately severe disability, unable to walk without assistance and unable to attend to own bodily needs without assistance; 5 = severe disability, bedridden, incontinent, and requiring constant nursing care and attention; and 6 = dead.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


