Kuru once affected many children ≥4 yr of age, adolescents, and young adults (mainly women) living in 1 limited area of Papua New Guinea. The complete disappearance of kuru among people born after 1957 suggests that the practice of ritual cannibalism (thought to have ended that year) was probably the only mechanism by which the infection was spread in Papua New Guinea.
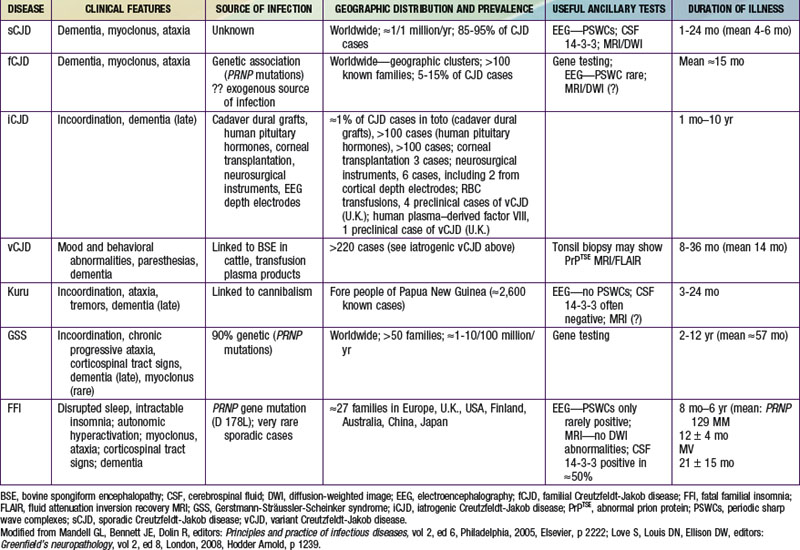
CJD, the most common human spongiform encephalopathy, was formerly thought to occur only in older adults; however, iCJD and, much more rarely, sCJD have affected adolescents and young adults. GSS and the insomnia syndromes have not been diagnosed in children or adolescents. Variant CJD has a peculiar predilection for younger people; of 174 cases of CJD reported to date in the U.K., all except 23 were in people younger than 40 yr of age and 22 were under 20 yr of age. CJD has been recognized worldwide, at yearly rates of 0.25 to 2 cases/million population (not age-adjusted), with foci of considerably higher incidence among Libyan Jews in Israel, in isolated villages of Slovakia, and in other limited areas. Sporadic CJD has not been convincingly linked to any common exposure, and the source of infection remains unknown. Epidemiologic surveys have investigated several hypothetical mechanisms of spread of CJD. Person-to-person spread has been confirmed only for iatrogenic cases. Spouses and household contacts of patients are at very low risk of acquiring CJD, although 2 instances of conjugal CJD have been reported. However, medical personnel exposed to brains of patients with CJD may be at some increased risk; at least 20 health care workers have been recognized with the disease.
The striking resemblance of CJD to scrapie prompted a concern that infected sheep tissues might be a source of spongiform encephalopathy in humans. No reliable epidemiologic evidence suggests that exposure to potentially scrapie-contaminated animals, meat, meat products, or experimental preparations of the scrapie agent have transmitted a TSE to humans. The potential of the CWD agent to infect human beings has not been demonstrated but remains under investigation; deer, elk, and moose in 16 U.S. states and 2 Canadian provinces have been naturally infected and monkeys have been experimentally infected with the CWD agent. Exposure to contaminated meat, including venison from animals infected with the CWD agent, has not been implicated as a risk factor for sporadic CJD.
The outbreak of BSE among cattle (possibly infected by eating scrapie-agent–contaminated meat-and-bone meal added to feed) was 1st recognized in the United Kingdom in 1986, later reported in native cattle of 24 other countries, including Canada and the USA. The finding of a new TSE in ungulate and feline animals in British zoos and later in domestic cats raised a fear that some TSE agent (probably a strain of the scrapie agent), having crossed the species barrier from sheep to cattle, had acquired a broadened range of susceptible hosts, posing a potential danger for humans. That is the plausible explanation for the occurrence of vCJD, 1st described in adolescents in Britain in 1996 and as of December 2010 affecting at least 174 people in the U.K. (not counting several with evidence of “preclinical” vCJD infection), 25 in France, 5 in Spain, 4 in Ireland, 3 in the Netherlands, 2 each in Italy and Portugal, and single cases in Italy, Japan, and Saudi Arabia. Variant CJD has also occurred in former U.K. residents living in Canada (1 case) and the USA (2 cases); a 3rd case of vCJD in the USA was reported in a former resident of Saudi Arabia, a country that has not recognized BSE but might have imported contaminated meat products.
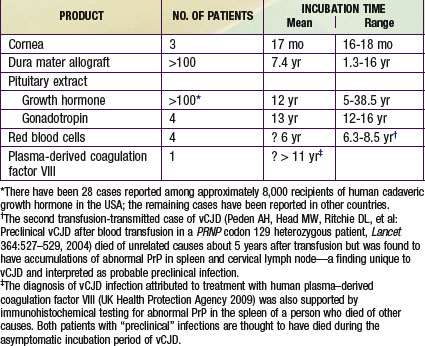
Iatrogenic transmissions of CJD have been recognized for >30 yr (Table 270-2). Such accidental transmissions of CJD have been attributed to use of contaminated neurosurgical instruments or operating facilities, use of cortical electrodes contaminated during epilepsy surgery, injections of human cadaveric pituitary growth hormone and gonadotropin, and transplantation of contaminated corneas and allografts of human dura mater used as a surgical patching material. Pharmaceuticals and tissue grafts derived from or contaminated with human neural tissues, particularly when obtained from unselected donors and large pools of donors, pose special risks.
Only gold members can continue reading.
Log In or
Register to continue
Related
 on the Nelson Textbook of Pediatrics website at www.expertconsult.com): kuru; Creutzfeldt-Jakob disease (CJD) with its variants—sporadic CJD (sCJD), familial CJD (fCJD), iatrogenic CJD (iCJD), and new-variant or variant CJD (vCJD); Gerstmann-Sträussler-Scheinker syndrome (GSS); and fatal familial insomnia (FFI), or the even more rare sporadic fatal insomnia syndrome. TSEs also affect animals; the most common and best-known TSEs of animals are scrapie in sheep, bovine spongiform encephalopathy (BSE or mad cow disease) in cattle, and a chronic wasting disease (CWD) of deer, elk, and moose found in parts of the USA and Canada. All TSEs have similar clinical manifestations and histopathology, and all are “slow” infections with very long asymptomatic incubation periods (often years), durations of several months or more, and overt disease affecting only the nervous system. The most striking neuropathologic change that occurs in each TSE, to a greater or lesser extent, is spongy degeneration of the cerebral cortical gray matter.
on the Nelson Textbook of Pediatrics website at www.expertconsult.com): kuru; Creutzfeldt-Jakob disease (CJD) with its variants—sporadic CJD (sCJD), familial CJD (fCJD), iatrogenic CJD (iCJD), and new-variant or variant CJD (vCJD); Gerstmann-Sträussler-Scheinker syndrome (GSS); and fatal familial insomnia (FFI), or the even more rare sporadic fatal insomnia syndrome. TSEs also affect animals; the most common and best-known TSEs of animals are scrapie in sheep, bovine spongiform encephalopathy (BSE or mad cow disease) in cattle, and a chronic wasting disease (CWD) of deer, elk, and moose found in parts of the USA and Canada. All TSEs have similar clinical manifestations and histopathology, and all are “slow” infections with very long asymptomatic incubation periods (often years), durations of several months or more, and overt disease affecting only the nervous system. The most striking neuropathologic change that occurs in each TSE, to a greater or lesser extent, is spongy degeneration of the cerebral cortical gray matter.