Thyroid Hormones
Delbert A. Fisher
Thyroid hormones are critically important to normal growth and development during infancy, childhood, and adolescence. Thyroid hormone deficiency during fetal life and infancy leads to mental retardation, and hypothyroidism during childhood and adolescence leads to growth and developmental retardation. In addition, there are significant effects of thyroid hormones on energy metabolism and on the metabolism of nutrients and inorganic ions, and these actions are important in the maintenance of normal metabolic homeostasis. In the present chapter, we review normal thyroid hormone physiology and pharmacology and the pharmacology of the drugs and chemicals used in the diagnosis and management of disorders of thyroid function.
Thyroid Hormone Synthesis and Release
Thyroid hormones and analogues are tyrosine derivatives. Their structures are summarized in Figure 53.1. The only source of thyroxine (tetraiodothyronine, T4) is the thyroid gland. The major substrates for T4 synthesis are iodide and tyrosine (1,2). Tyrosine is not rate limiting, but iodine is a trace element, the limitation of which can severely impair thyroid hormone synthesis. The steps in thyroid hormone synthesis by the thyroid gland include iodide trapping, synthesis of thyroglobulin (Tg), organification of trapped iodide, storage of iodinated Tg in follicular colloid, endocytosis and hydrolysis of Tg to release thyroid hormones, and deiodination of monoiodotyrosine (MIT) and diiodotyrosine (DIT) with intrathyroidal recycling of the iodide (1,2) (see Fig. 53.2).
The transport of iodide across the thyroid cell membrane is the first and rate-limiting step in thyroid hormone biosynthesis. The salivary glands, gastric mucosa, uterus, mammary glands, small intestine, and placenta also are able to concentrate iodide; however, they are not capable of iodothyronine synthesis. Normally, the thyroid follicular cell generates a thyroid/serum concentration gradient of 30- to 40-fold. This gradient increases markedly when stimulated by iodine deficiency, thyroid-stimulating hormone (thyrotropin; TSH), or thyroid-stimulating immunoglobulins (TSIs), or by drugs that impair the efficiency of hormone synthesis. Several inorganic anions are capable of competitively inhibiting iodide transport; these include bromide (Br), nitrate (NO2), thiocyanate (SCN), and perchlorate (CIO4).
The predominant protein in thyroid colloid is Tg, a large 660-kd homodimer. The oxidation of iodide to an active intermediate is followed by iodination of Tg-bound tyrosyl residues. The thyroid gland normally contains 50 to 100 mg Tg for every 1 g of gland. The tyrosyl residues, which are the iodine acceptors of Tg, comprise about 3% of the weight of the protein, and about two-thirds of these are spatially oriented to be susceptible to iodination. Iodination of Tg tyrosyl residues forms MIT and DIT. These iodotyrosines then couple to form the iodothyronines T3 and T4; DIT + DIT couple to form T4 while MIT + DIT coupling forms T3. The relative proportions of T3 and T4 formed depend on the amount of available iodide. About 30% of the Tg iodoprotein is iodothyronine, with a T4/T3 ratio of 10:1 to 20:1.
The first step in thyroid hormone release is the endocytosis of stored colloid. The ingested colloid droplets fuse with proteolytic enzyme-containing lysosomes to form phagolysosomes in which Tg is digested to release free iodothyronines into the cytoplasm for diffusion into blood (Fig. 53.2). The MIT and DIT released during hydrolysis of Tg are largely deiodinated by a thyroidal iodotyrosine deiodinase (1,2). The free iodide enters the intracellular iodide pool and is reutilized for new hormone synthesis (Fig. 53.2). A defect in thyroidal iodotyrosine deiodinase leads to a release of iodotyrosines into the circulation and their excretion in the urine. The loss from the thyroid gland of this normally recycled iodine, amounting to 70% to 80% of the daily thyroidal iodine supply, can significantly compromise thyroid hormone synthesis.
Some Tg is released into the circulation or the perivascular compartment during the process of synthesis, and there is good evidence that Tg reaches the general circulation via thyroidal lymphatics. Circulating Tg levels are high in premature infants during the first weeks of life, and values decrease with age throughout infancy and childhood (2,3). Circulating Tg concentrations in normal children range from 2 to 70 ng per mL. Values increase after TSH administration and decrease during thyroid hormone administration.
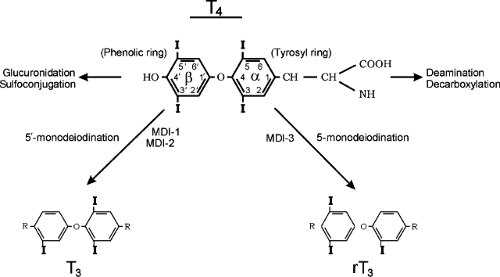 Figure 53.1. The metabolism of thyroxine (tetraiodothyronine). The major metabolic pathway is progressive monodeiodination by the three iodothyronine deiodinase enzymes, MDI-1, MDI-2, and MDI-3. Outer (phenolic) ring 5′-monodeiodination produces active 3,5,3′-triiodothyronine. Inner (tyrosyl) ring 5-monodeiodination produces inactive reverse (3,3′,5′) triiodothyronine. MDI-1 is also capable of inner ring monodeiodination. The alanine side chain of the tyrosyl ring is subject to deamination and decarboxylation. Sulfoconjugation and glucuronide conjugation reactions at the 4′-phenolic ring site occur in liver tissue. |
Regulation of Thyroid Function
Thyroid follicular cell function is regulated largely by circulating TSH and iodide levels (1,2,4). TSH stimulates production of intracellular cyclic adenosine monophosphate (cAMP), and cAMP appears to mediate most of the effects of TSH on thyroid metabolism. Pituitary TSH secretion is modulated by hypothalamic thyrotropin-releasing hormone (TRH) and negative feedback action of thyroid hormones. TRH stimulates TSH release and increases TSH synthesis as well as TSH glycosylation. Feedback control is mediated via pituitary cell T3 nuclear receptors that modulate TSH synthesis and pituitary cell membrane TRH-receptor binding. Increased thyroid hormone levels inhibit and decreased concentrations stimulate pituitary TSH synthesis. The rate of pituitary TSH release is the net effect of the stimulation by TRH and the inhibitory effect of T3 (see Fig. 53.3). Hypothalamic TRH production is modulated by environmental temperature via both peripheral and central (hypothalamic) thermal receptors. Decreasing environmental and body temperatures increases TRH production and increases the tonic level of TSH release. Thyroid hormones also modulate TRH synthesis within the hypothalamus.
The TRH neurons in the paraventricular hypothalamic nucleus receive input from other regions of the brain and from the circulation (4). The major afferent links to these neurons include catecholamine neurons in the brain stem and neurons from the arcuate nucleus. The former likely mediates upregulation of the TRH gene during cold exposure, while the latter mediates downregulation during starvation and illness (4). Administration of the adipocyte hormone leptin, the production of which falls during fasting, prevents the fasting-induced fall in TSH. In addition, central administration of α-melanocyte-stimulating hormone (α-MSH) prevents the fasting-induced suppression of TSH. Agouti-related peptide and neuropeptide are orexigenic peptides that are downregulated by leptin and upregulated during fasting. When administered centrally, both can suppress TRH and TSH secretion. Thus, the arcuate nucleus integrates leptin signaling and assists in the regulation of TRH gene expression as well as food intake and energy expenditure (4).
The average level of plasma iodide also is an important factor in the control of thyroid gland function (1,2). Variation in iodine intake in the physiologic range regulates thyroid membrane iodide trapping, and in pharmacologic doses iodide will block thyroid hormone synthesis. At least one important mechanism for these effects is the inhibitory action of iodide on the stimulation of cAMP by TSH.
Metabolism of Thyroid Hormones
The thyroid gland is the sole source of T4, but most of the T3 in the blood is derived from nonglandular sources via monodeiodination of T4 in peripheral tissues (1,2) (see Fig. 53.3). Both T3 and T4 in the blood are associated with plasma proteins; the binding affinities of these proteins are greater for T4 than T3 and the concentration of T4 in human blood is 50 to 100 times greater than that of T3 (5). The concentrations of both are relatively constant in the
steady state. The plasma half-life of T4 in adult humans approximates 5 days; the half-life of T3 approximates 1 day. The circulating thyroid hormone-binding proteins include T4-binding inter-alpha globulin (TBG), T4-binding prealbumin (transthyretin), and albumin. TBG is the most important carrier protein for T4; TBG and albumin seem equally important for T3. The euthyroid steady-state concentrations of free T4 and free T3 approximate 0.02% and 0.20%, respectively, of the total hormone concentrations. Absolute mean free T4 and T3 concentrations approximate 1.5 and 0.3 ng per dL, respectively. In adolescents and adults, the plasma concentrations of the several binding proteins are 1 to 4 mg per dL for TBG, 10 to 20 mg per dL for transthyretin, and 2 to 5 g per dL for albumin. TBG levels are higher in children than in adults and decrease progressively to adult levels during adolescence.
steady state. The plasma half-life of T4 in adult humans approximates 5 days; the half-life of T3 approximates 1 day. The circulating thyroid hormone-binding proteins include T4-binding inter-alpha globulin (TBG), T4-binding prealbumin (transthyretin), and albumin. TBG is the most important carrier protein for T4; TBG and albumin seem equally important for T3. The euthyroid steady-state concentrations of free T4 and free T3 approximate 0.02% and 0.20%, respectively, of the total hormone concentrations. Absolute mean free T4 and T3 concentrations approximate 1.5 and 0.3 ng per dL, respectively. In adolescents and adults, the plasma concentrations of the several binding proteins are 1 to 4 mg per dL for TBG, 10 to 20 mg per dL for transthyretin, and 2 to 5 g per dL for albumin. TBG levels are higher in children than in adults and decrease progressively to adult levels during adolescence.
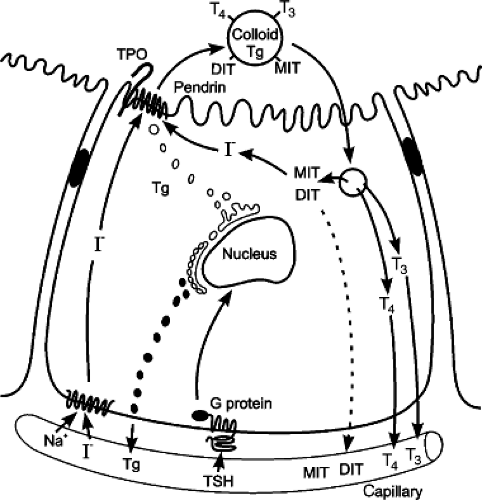 Figure 53.2. Drawing of the thyroid follicular cell illustrating thyroid hormone synthesis and secretion. TSH regulates the process through the G protein-linked plasma membrane TSH receptor. TSH binding stimulates thyroglobulin (Tg) synthesis and sodium-iodide symporter uptake of circulating iodide. Small amounts of Tg are secreted, presumably from the endoplasmic reticulum into the thyroid lymphatics. The tyrosine residues of thyroglobulin are iodinated at the apical cell membrane catalyzed by thyroid peroxidase, the organification enzyme. The pendrin protein, a chloride/iodide transporter located at the apical cell membrane, facilitates organification. The resulting monoiodotyrosine (MIT), and diiodotyrosine (DIT) moieties with appropriate spatial orientation couple to form thyroxine (T4) and triiodothyronine (T3) within the stored thyroglobulin molecule. TSH also stimulates endocytosis of colloid droplets and progressive thyroglobulin proteolysis within the resulting phagolysosomes. The released T4 and T3 are secreted into the circulation. The uncoupled MIT and DIT are deiodinated by iodotyrosine deiodinase to release iodide, which is largely recycled within the follicular cell. |
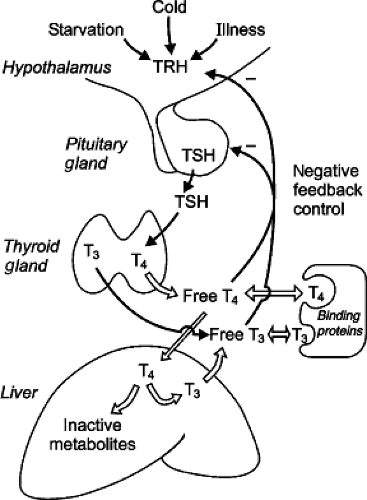 Figure 53.3. Drawing of the hypothalamic–pituitary–thyroid axis. Hypothalamic TRH stimulates pituitary TSH production, which increases thyroid follicular cell activity and thyroid hormone secretion. Most T3 is produced in peripheral tissues (here shown as liver) from T4 catalyzed by iodothyronine deiodinase enzymes. The deiodination process produces approximately similar amounts of T3 and reverse (inactive) T3 or rT3 (see Fig. 53.1). Most circulating T4 and T3 are protein bound. The free fractions act via a negative feedback loop to inhibit TRH and TSH maintaining serum free hormone levels within a narrow physiologic range. TSH secretion is regulated by negative feedback of thyroid hormones on hypothalamic TRH secretion and pituitary TSH synthesis and secretion. This feedback is mediated via TRβ thyroid receptors. Cold exposure, starvation or fasting, and nonthyroidal illness also modulate TRH and TSH secretion. See text for details. |
Two major extravascular kinetic pools of T3 and T4 probably exist, one in which plasma–tissue interchange is rapid (chiefly liver, kidney, and lung) and one in which exchange is slow (chiefly skeletal, muscle, and skin) (6). An additional pool, chiefly in gut and bone, with an intermediate exchange rate has been suggested. Peak T4 concentrations after single-pulse doses of labeled hormone occur in these “pools” in minutes, hours, and days, respectively. Total body T4 distribution in adults has been estimated as follows: about 20% in plasma, 30% in fast tissues, 45% in slow tissues, and 5% in intermediate tissues.
Deiodination is the major pathway of thyroid hormone metabolism in humans. The first step in T4 metabolism is conversion either to T3 or to reverse T3 (1,2,7) (Fig. 53.1). Monodeiodination of the β or outer (hydroxyl) ring produces T3, which has three to four times the metabolic potency of T4. Monodeiodination of the α or inner ring produces rT3, which is metabolically inactive. Two types of outer ring iodothyronine monodeiodinase have been described (Fig. 53.1). MDI-1, predominantly expressed in liver, kidney, and thyroid, is a high Km enzyme,
inhibited by propylthiouracil (PTU), and stimulated by thyroid hormone. MDI-2 also is capable of inner ring monodeiodination, particularly of T4 sulfate (to rT3 sulfate) and T3 sulfate (to T2 sulfate) (7,8). MDI-2, predominantly located in brain, pituitary, placenta, skeletal muscle, heart, thyroid, and brown adipose tissues (BATs), is a low Km enzyme insensitive to PTU and inhibited by thyroid hormone. Both MDI-1 and MDI-2 contribute to circulating T3 production while MDI-2 acts as well to increase local tissue levels of T3 (7,8). A third inner (tyrosyl) ring iodothyronine monodeiodinase (MDI-3) has been characterized in most tissues, including brain and placenta and more recently in uterus and fetal skin. This enzyme system catalyzes the conversion of T4 to rT3 and T3 to diiodothyronine.
inhibited by propylthiouracil (PTU), and stimulated by thyroid hormone. MDI-2 also is capable of inner ring monodeiodination, particularly of T4 sulfate (to rT3 sulfate) and T3 sulfate (to T2 sulfate) (7,8). MDI-2, predominantly located in brain, pituitary, placenta, skeletal muscle, heart, thyroid, and brown adipose tissues (BATs), is a low Km enzyme insensitive to PTU and inhibited by thyroid hormone. Both MDI-1 and MDI-2 contribute to circulating T3 production while MDI-2 acts as well to increase local tissue levels of T3 (7,8). A third inner (tyrosyl) ring iodothyronine monodeiodinase (MDI-3) has been characterized in most tissues, including brain and placenta and more recently in uterus and fetal skin. This enzyme system catalyzes the conversion of T4 to rT3 and T3 to diiodothyronine.
From 70% to 90% of circulating T3 is derived from peripheral conversion and 10% to 25% from the thyroid gland; relative values for reverse T3 are 96% to 98% and 2% to 4%. Progressive tissue monodeiodination reactions degrade T3 and rT3 to diiodo-, monoiodo-, and noniodinated thyronine. Conjugation of the phenolic ring hydroxyl group to sulfate or glucuronide produces glucuronide or sulfate analogues of the iodothyronines (7,8). These are inactivating reactions because the analogues formed do not bind to thyroid hormone receptors and are rapidly metabolized by MDI-1 or excreted in bile (7,8). The exception is T3 sulfate which demonstrates some 20% of native T3 bioactivity when injected into hypothyroid rats. This presumably is due to the action of tissue sulfatases or bacterial sulfatases in the intestine (8).
The alanine side chain of the inner ring of the hormones also is subject to degradative reactions, including deamination, and decarboxylation (7,8). Pyruvic acid analogues and small amounts of a lactic acid analogues have been observed in urine and bile; these have minimal biologic activity. Two enzymes, L-amino acid oxidase and thyroid hormone aminotransferase, catalyze the deamination of T4 and T3 to their acetic acid analogues tetrac and triac (7,8). Triac binds to the T3 nuclear receptor with greater avidity than T3 and demonstrates significant biologic activity when injected into animals or humans. However, relatively large doses are required because of rapid clearance and degradation via deiodination and glucuronidation (7,8). Triac has been utilized in the treatment of thyroid hormone resistance. T4 and T3 are potent proangiogenic agents in the chick chorioallantoic membrane model of angiogenesis and in human dermal microvascular endothelial cell assays (9). These actions are mediated via a plasma membrane hormone receptor site on integrin αv β3. Tetrac has been shown to block this binding and to inhibit thyroid hormone action on tumor cell proliferation. Tetrac has no agonist activity at the hormone receptor site (9).
Recently, two novel thyroid hormone derivatives, 3-iodothyronamine (T1AM) and thyronamine (T0AM), products of combined thyroid hormone decarboxylation and deiodination, have been shown in mice to induce transient hypothermia, decrease aortic pressure, block carbohydrate utilization, and increase fat utilization (10,11). These molecules are present in mammalian brain, peripheral organs, thyroid gland, and blood serum. T1AM is strongly protein bound and circulating concentrations are in the microgram per deciliter range similar to T4 (12). It has been proposed that these thionamines act via a thyronamine G-protein-coupled receptor such as the trace amine-associated receptor (TAAR1), a 7-transmembrane, G-protein-coupled receptor related to catecholamine and 5-hydroxytryptophan receptors (10). Their physiologic and clinical significance remain unclear.
Thyroid Hormone Actions
Thyroid hormone effects are mediated predominantly by means of nuclear thyroid hormone protein receptors (TR), which act as DNA binding transcription factors regulating gene transcription. Two mammalian genes code for TR, Trα, and TRβ, and alternative splicing of expressed mRNA species leads to production of the major isoforms Trα1, Trα2, TRβ1, and TRβ2 (13,14,15,16). In rodent species, hepatic TR-binding activity matures during the first 3 to 5 weeks of extrauterine life. Trα binding in brain and pituitary cells appears during fetal life, whereas TRβ isoforms increase during the early neonatal period (15). In the fetal/neonatal rat, thyroid hormone effects on thermogenesis, hepatic enzyme activities, skin and brain maturation, and growth hormone (GH) metabolism mature largely during the first 4 postnatal weeks (16,17).
In the human fetus, low levels of T4 binding have been detected in brain tissue at 10 weeks of gestational age, and hepatic, cardiac, and lung TR binding are observed at 16 to 18 weeks (16,18). Information is limited regarding the timing of appearance of thyroid hormone postreceptor effects in the human fetus. The length, weight, appearance, behavior, biochemical parameters, extrauterine adaptation, and early neonatal course of the athyroid human neonate usually are normal (16,18). Growth of the human fetus is programmed independent of thyroid hormones by a complex interplay of genetic, nutritional, hormonal, and growth factors.
Postnatal thermogenesis is mediated through BAT, prominent in subscapular and perirenal areas in the mammalian fetus and neonate. Heat production in BAT is stimulated by catecholamines by means of β-adrenergic receptors and is thyroid hormone dependent. The uncoupling protein thermogenin, or UCP-1, unique to BAT, is located on the inner mitochondrial membrane and uncouples phosphorylation by dissipating the proton gradient created by the mitochondrial respiratory chain. An iodothyronine deiodinase (MDI) in BAT mediates local T4 to T3 conversion. Full thermogenin expression in BAT requires both catecholamine and T3 stimulation (19,20). UCP-2 is found in many tissues but does not appear to be regulated by β-adrenergic agonists or thyroid hormone. UCP-3 has been cloned and is expressed in muscle and white adipose tissue as well as BAT (20). Muscle UCP-3 is regulated by β3-adrenergic stimulation and thyroid hormone and probably contributes to nonshivering thermogenesis in adult rats and presumably humans. UCP-3 mRNA levels also are regulated by dexamethasone, leptin, and starvation, but the regulation differs in BAT and muscle (20). TR-mediated thyroid hormone actions also include the synthesis of selected enzymes and proteins in liver; stimulation of myogenin, α-actin, and myosin synthesis in cardiac tissue; promotion of mammary gland development and gene expression; and stimulation of growth and development and central nervous system (CNS) maturation.
Studies in the rat model have characterized a profound effect of thyroid hormones on GH secretion and action (15,21). Hypothyroidism decreases pituitary GH content, impairs the pituitary GH response to GH-releasing hormone (GHRH), reduces basal and pulsatile GH secretion, and decreases circulating GH levels. Similar effects have been observed in hypothyroid human subjects. Hypothyroid patients show limited GH responses to insulin hypoglycemia, arginine, and GHRH, reduced nocturnal GH secretion, and reduced circulating levels of insulin-like growth factor I, (IGFI), IGF-II, IGF-binding protein-3, GH-binding protein, and bioactive IGF (22,23,24). There is also evidence that thyroid hormones stimulate production of other growth factors, including epidermal growth factor, nerve growth factor, and erythropoietin and have direct actions on bone, potentiating the cartilage response to IGF-I, and stimulating osteoblastic bone resorption and remodeling (25,26,27).
These hormonal and growth factor deficiencies contribute importantly to the disordered growth of hypothyroid infants and children, including the decreased long-bone growth, delayed bone maturation, decreased carcass growth, delayed tooth eruption, and anemia. The actions of thyroid hormones and GH are synergistic. GH administered to neonatal hypophysectomized rats increases body weight, but it has a limited effect on skeletal growth; in contrast, thyroxine accelerates skeletal growth and potentiates the GH effects. Combined treatment optimizes growth and development (21). The vast majority of infants with congenital hypothyroidism (CH) are born with normal or increased length and weight, and early adequate thyroid hormone treatment prevents development of growth retardation. There may be a transient period of growth deceleration during the early weeks of treatment (27), but even in infants with severe CH, manifested by very low serum T4 concentrations and delayed bone maturation at birth, early, adequate treatment results in mean height and body mass index equal to or greater than values in normal children (28,29,30,31). Bone age values also are normalized by 2 to 3 years (32,33). Bone mineral density and metabolism also are normal in treated children given adequate nutrition (34).
The effects of thyroid hormones on growth and physical development extend through most of the second decade, and delayed or inadequate replacement therapy during childhood or adolescence can reduce adult height (35). Thyroxine treatment in children with growth retardation due to a prolonged period of untreated hypothyroidism usually induces marked catch-up growth, but in some instances this is inadequate to normalize adult height (35,36). Adult height is generally inversely correlated with the duration of untreated hypothyroidism prior to the advent of puberty (35). This is thought to be due to the onset of puberty, limiting the chance to achieve full growth catch-up before epiphyseal fusion. Suppression of pubertal development by administration of gonadotropin-releasing hormone plus GH was shown to improve height gain in a juvenile hypothyroid patient (35).
The critical role of thyroid hormones in CNS maturation has long been recognized. The first phase of fetal CNS maturation (neuronal multiplication, migration, and organization) occurs during the second trimester of gestation. A second phase (glial cell multiplication, migration, and myelinization) occurs during the perinatal period and extends through 3 to 4 postnatal year (16,18). Available evidence suggests that deficiency or excess of thyroid hormones alters the timing or synchronization of the CNS developmental program, presumably by altering critical homeobox gene cascades and other genetic CNS maturation events (16,18). Prior to activation of fetal thyroid function by the fetal hypothalamic–pituitary complex at midgestation, the fetus is dependent on maternal thyroid hormone which traverses the placenta in limited amounts. Several studies have shown that maternal hypothyroidism, often due to Hashimoto autoimmune thyroiditis, impairs the subsequent neuropsychological development of the offspring; even mild maternal hypothyroidism has been shown to reduce offspring IQ by 4 to 7 points (36,37).
Severe iodine deficiency with combined maternal and fetal hypothyroidism results in severe neurological impairment (cretinism), and mild endemic iodine deficiency, producing mild maternal and childhood hypothyroxinemia, has been shown to reduce the mean IQ of children (37). Sixty percent to 70% of postnatal brain growth and differentiation occurs during the first 2 years of life, and it is during this period that the CNS effects of hypothyroidism are most devastating (38). Untreated, severe CH is associated with an IQ deficit of 4 to 6 points per month during the first 5 to 7 months of postnatal life (17,37). Such infants treated promptly at birth have minimal IQ impairment, being protected in utero by the limited amounts of maternal thyroxine crossing the placenta (39,40,41,42). However, overall, a mean 6-point IQ deficit has been shown in a meta-analysis of published treatment outcome papers (40). This deficit is largely due to infants with severe disease with intrauterine hypothyroidism manifest as delayed bone maturation at birth (41,42). These data, summarized in Table 53.1, emphasize the critical role of
thyroid hormones in CNS development both in utero and during infancy and early childhood.
thyroid hormones in CNS development both in utero and during infancy and early childhood.
Table 53.1 Deficiency Relative to Phase of Central Nervous System Development | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||
Relative Effects of Thyroid Hormones Versus Age
Thyroid hormone actions vary with age. Thyroid hormone deficiency during human fetal life has minimal untoward effects. Somatic growth and development and linear bone growth proceed normally in the athyroid fetus, and bone maturation is normal or minimally retarded (3 to 6 weeks) at birth (43,44,45). The reason(s) for the relative lack of thyroid hormone effect on fetal somatic growth is not clear. Likely explanations include low levels of active thyroid hormone in fetal serum and tissues and/or immaturity of thyroid hormone receptor responsiveness at the transcription, translation, or action levels (17,46,47).
The developmental effects of thyroid hormones, such as the CNS effects, are most obvious during infancy and early childhood. Somatic growth, bone growth and maturation, and tooth development and eruption are thyroid dependent. After 3 to 4 years of age, thyroid hormone deficiency is not associated with mental retardation, but delayed somatic and linear bone growth and delayed eruption of permanent dentition are prominent. Bone maturation, measured as bone age, also is delayed, diaphyseal bone growth is reduced, and epiphyseal growth and mineralization largely cease (2,27).
Hypothalamic and anterior pituitary function also may be abnormal in hypothyroid children. Although in most children thyroid hormone deficiency leads to a delayed sexual development, occasional children manifest precocious sexual maturation with increased levels of circulating gonadotropins (48,49,50). In female patients, serum prolactin levels also tend to be increased, and galactorrhea may occur if serum estrogen levels are high enough to permit breast development and milk production. These changes seem to occur in children with high serum TSH levels, and enlargement of the sella turcica has been observed. The increased serum prolactin levels are probably explained by the fact that TRH stimulates both TSH and prolactin release from the pituitary. A paracrine action of the hyperstimulated thyrotropic cells on gonadotrope cells may explain the increased gonadotropin secretion. Precocious puberty is observed only rarely, so it is possible that these patients have genetic variants of the gonadotropin receptors, which can be stimulated by the increased TSH levels (51). Similar findings have been reported for TSH and follicle-stimulating hormone (FSH) receptor variants stimulated by βHCG (51,52). When the hypothyroid state is alleviated, the manifestations of sexual precocity regress, and normal puberty ensues when the general level of maturity has progressed appropriately.
During childhood and adolescence and until epiphyseal closure, thyroid hormone deficiency leads to reduced somatic growth, reduced linear bone growth, and delayed bone maturation. In addition, epiphyseal dysgenesis is commonly observed (27,53). The effects of thyroid hormone deficiency on dental development are less profound, but delay in eruption of second dentition may occur. Abnormalities of hypothalamic–pituitary function secondary to hypothyroidism also are common in adolescence (2,27). Puberty often is delayed or incomplete. In normal women, menstrual cycles commonly are nonovulatory and bleeding may be irregular. This pattern usually is more prolonged in hypothyroid female adolescents. In addition, menorrhagia or hypomenorrhea may occur.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


