Chapter 489 The Leukemias
489.1 Acute Lymphoblastic Leukemia
Etiology
In virtually all cases, the etiology of ALL is unknown, although several genetic and environmental factors are associated with childhood leukemia (Table 489-1). Exposure to medical diagnostic radiation both in utero and in childhood has been associated with an increased incidence of ALL. In addition, published descriptions and investigations of geographic clusters of cases have raised concern that environmental factors can increase the incidence of ALL. Thus far, no such factors other than radiation have been identified in the USA. In certain developing countries, there has been an association between B-cell ALL and Epstein-Barr viral infections.
Cellular Classification
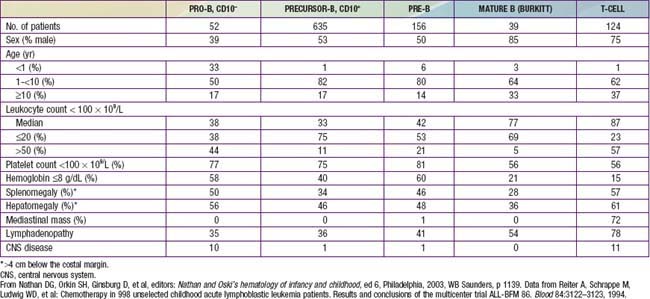
The classification of ALL depends on characterizing the malignant cells in the bone marrow to determine the morphology, phenotypic characteristics as measured by cell membrane markers, and cytogenetic and molecular genetic features. Morphology alone usually is adequate to establish a diagnosis, but the other studies are essential for disease classification, which can have a major influence on the prognosis and the choice of appropriate therapy. The most important distinguishing morphologic feature is the French-American-British (FAB) L3 subtype, which is evidence of a mature B-cell leukemia. The L3 type, also known as Burkitt leukemia, is one of the most rapidly growing cancers in humans and requires a different therapeutic approach than other subtypes of ALL. Phenotypically, surface markers show that about 85% of cases of ALL are derived from progenitors of B cells, about 15% are derived from T cells, and about 1% are derived from B cells. A small percentage of children with leukemia have a disease characterized by surface markers of both lymphoid and myeloid derivation. Immunophenotypes often correlate to disease manifestations (Table 489-2).
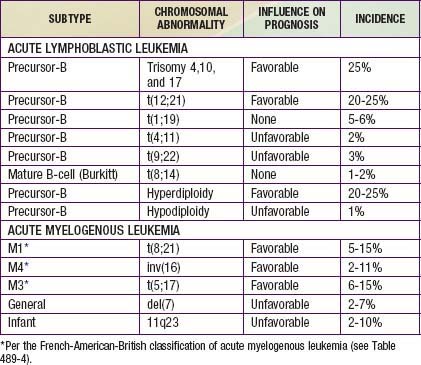
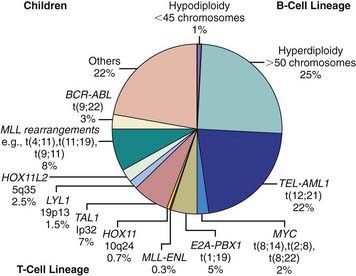
Chromosomal and genetic abnormalities are found in most patients with ALL (Table 489-3, Fig. 489-1). The abnormalities, which may be related to chromosomal number, translocations, or deletions, provide important prognostic information. The identification of the leukemia-specific fusion-gene sequences in archived neonatal blood spots of some children who develop ALL at a later date indicates the importance of in utero events in the initiation of the malignant process, but the long lag period before the onset of the disease in some children, reported to be as long as 14 yr, supports the concept that additional genetic modifications also are required for disease expression. The polymerase chain reaction and fluorescence in situ hybridization techniques offer the ability to pinpoint molecular genetic abnormalities and to detect small numbers of malignant cells during follow-up and are of proven clinical utility. The development of DNA microanalysis makes it possible to analyze the expression of thousands of genes in the leukemic cell. This technique promises to further enhance the understanding of the fundamental biology and to provide clues to the therapeutic approach of ALL. Some effectors of critical signal transduction pathways have already been implicated in the pathogenesis of ALL using this technique.
Clinical Manifestations
On physical examination, findings of pallor, listlessness, purpuric and petechial skin lesions, or mucous membrane hemorrhage can reflect bone marrow failure (Chapter 487). The proliferative nature of the disease may be manifested as lymphadenopathy, splenomegaly, or, less commonly, hepatomegaly. In patients with bone or joint pain, there may be exquisite tenderness over the bone or objective evidence of joint swelling and effusion. Nonetheless, with marrow involvement, deep bone pain may be present but tenderness will not be elicited. Rarely, patients show signs of increased intracranial pressure that indicate leukemic involvement of the CNS. These include papilledema (see Fig. 487-3), retinal hemorrhages, and cranial nerve palsies. Respiratory distress usually is related to anemia but can occur in patients with an obstructive airway problem (wheezing) due to a large anterior mediastinal mass (e.g., in the thymus or nodes). This problem is most typically seen in adolescent boys with T-cell ALL. T-cell ALL also has a higher leukocyte count.
Precursor B-cell ALL (CD10+ or common acute lymphoblastic leukemia antigen [CALLA] positive) is the most common immunophenotype (see Table 489-2), with onset at 1-10 yr of age. The median leukocyte count at presentation is 33,000, although 75% of patients have counts <20,000; thrombocytopenia is seen in 75% of patients, and hepatosplenomegaly is seen in 30-40% of patients. In all types of leukemia, CNS symptoms are seen at presentation in 5% of patients (5-10% have blasts in the CSF). Testicular involvement is rarely evident at diagnosis, but prior studies have indicated occult involvement in 25% of boys. There is no indication for testicular biopsy.
Treatment
The single most important prognostic factor in ALL is the treatment: Without effective therapy, the disease is fatal. The survival rates of children with ALL since the 1970s have improved as the results of clinical trials have improved the therapies and outcomes (Fig. 489-2). Survival is also related to age (Fig. 489-3) and subtype (Fig. 489-4).
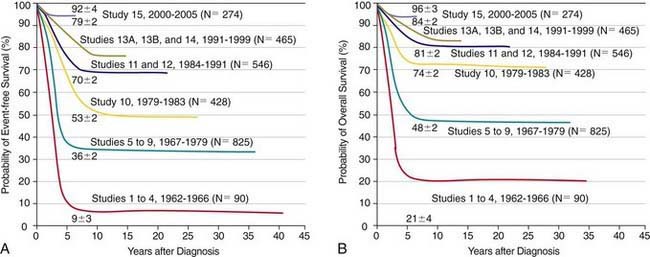
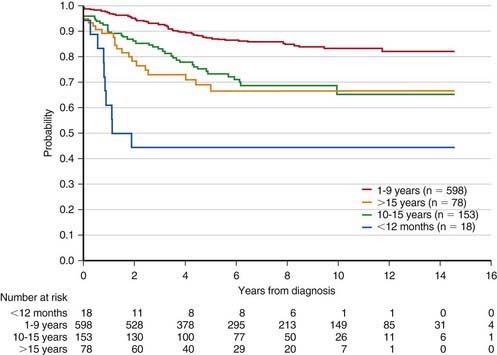
Figure 489-3 Kaplan-Meier estimates of event-free survival according to age at diagnosis of acute lymphoblastic leukemia.
(From Pui CH, Robinson LL, Look AT: Acute lymphoblastic leukaemia, Lancet 371:1030–1042, 2008.)
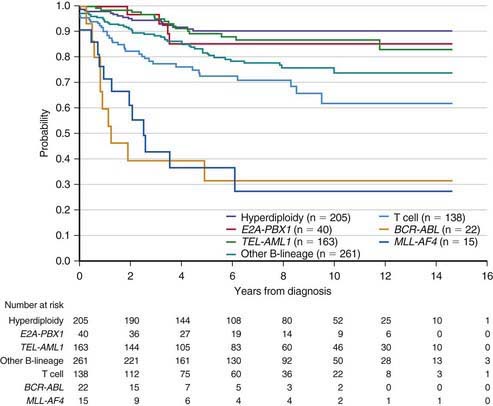
Figure 489-4 Kaplan-Meier analysis of event-free survival according to biological subtype of leukemia.
(From Pui CH, Robinson LL, Look AT: Acute lymphoblastic leukaemia, Lancet 371:1030–1042, 2008.)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


