Chapter 462 The Inherited Pancytopenias
Inherited (“constitutional”) pancytopenia is defined as a decrease in marrow production of the 3 major hematopoietic lineages that occurs on an inherited basis, resulting in anemia, neutropenia, and thrombocytopenia. Any of these conditions (Table 462-1) can be transmitted as a simple mendelian disorder by mutant genes with inherited patterns of autosomal dominant, autosomal recessive, or X-linked types. Modifying genes and acquired factors may also be operative. Inherited pancytopenias account for approximately 30% of cases of pediatric marrow failure. Fanconi anemia is the most common of these disorders.
Fanconi Anemia
Clinical Manifestations
The most common anomaly in FA is hyperpigmentation of the trunk, neck, and intertriginous areas, as well as café-au-lait spots and vitiligo, alone or in combination (Fig. 462-1 and Table 462-2). Half the patients have short stature. Growth failure may be associated with abnormal growth hormone secretion or with hypothyroidism. Absence of radii and thumbs that are hypoplastic, supernumerary, bifid, or absent are common. Anomalies of the feet, congenital hip dislocation, and leg abnormalities are seen. A male patient with FA may have an underdeveloped penis; undescended, atrophic, or absence of the testes; and hypospadias or phimosis. Females can have malformations of the vagina, uterus, and ovary. Many patients have a FA “facies,” including microcephaly, small eyes, epicanthal folds, and abnormal shape, size, or positioning of the ears (see Fig. 462-1). Ectopic, pelvic, or horseshoe kidneys are detected by imaging and may show other organs as duplicated, hypoplastic, dysplastic, or absent kidneys. Cardiovascular and gastrointestinal malformations also occur. Approximately 10% of patients with FA are cognitively delayed.
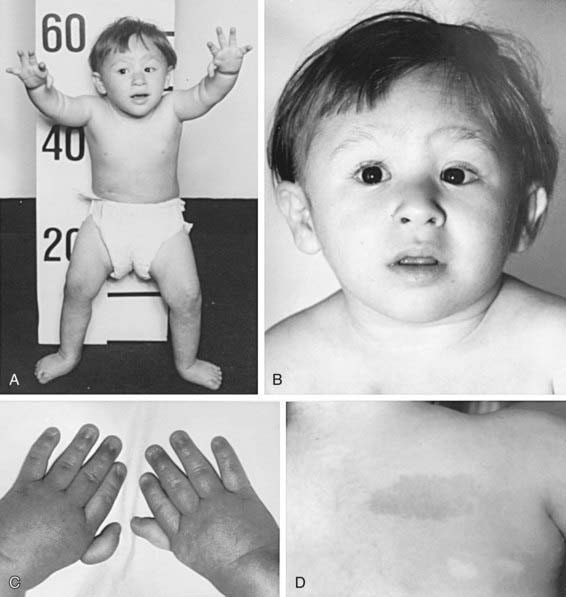
Table 462-2 CHARACTERISTIC PHYSICAL ANOMALIES IN FANCONI ANEMIA
| ANOMALY | APPROXIMATE FREQUENCY (% OF PATIENTS) |
|---|---|
| Skin pigment changes ± café-au-lait spots | 55 |
| Short stature | 51 |
| Upper limb abnormalities (thumbs, hands, radii, ulnas) | 43 |
| Hypogonadal and genital changes (mostly male) | 35 |
| Other skeletal findings (head/face, neck, spine) | 30 |
| Eye/lid/epicanthal fold anomalies | 23 |
| Renal malformations | 21 |
| Gastrointestinal/cardiopulmonary malformations | 11 |
| Hip, leg, foot, toe abnormalities | 10 |
| Ear anomalies (external and internal), deafness | 9 |
Treatment
Hematopoietic stem cell transplantation (HSCT; Chapter 129) is the only curative therapy for the hematologic abnormalities. Patients with FA <10 yr old who undergo transplantation using an HLA–identical sibling donor have a survival rate >80%. Survival rates are lower for patients undergoing the procedure when >10 yr. Preparative regimens are continuously evaluated, refined, and improved worldwide. For patients who do not have a matched sibling donor, a search for a matched unrelated donor (including a search of umbilical cord blood banks) might be initiated. Because of the heightened graft vs host response in patients with FA, the survival and cure rates have not been as good as those for matched sibling donor HSCT (≈ 30% survival). Molecular technology has led to preimplantation genetic diagnosis on parent-derived blastomeres to find an HLA-matched sibling donor without FA.



