I. Failure of formation of parts (arrest of development)
(A) Transverse deficiency
(B) Longitudinal deficiencies
(C) Phocomelia
(D) Tendon or muscle dysplasia
(E) Absent nail or skin
II. Failure of differentiation of parts
(A) Synostosis
(B) Radial head dislocation
(C) Symphalangism
(D) Contracture
(E) Tumorous conditions
III. Duplication
(A) Thumb polydactyly
(B) Central polydactyly
(C) Polydactyly of the little finger
(D) Opposable triphalangeal thumb
(E) Other types of hyperphalangism
(F) Mirror hand
IV. Abnormal induction of digital rays
(A) Soft tissue
(B) Bone
V. Overgrowth
(A) Macrodactyly
(B) Hemihypertrophy
VI. Undergrowth
(A) Microcheiria (hypoplastic hand)
(B) Brachydactyly
(C) Clinodactyly
VII. Constriction band syndrome
(A) Constriction ring
(B) Lymphedema
(C) Acrosyndactyly
(D) Amputation type
VIII. Generalized skeletal abnormalities and a part of syndrome
IX. Others (including unclassifiable cases)
Table 2
OMT classification of the congenital anomalies of the hand and upper extremity (2010)
1. Malformations |
A. Failure of axis formation/differentiation – entire upper limb |
1. Proximal-distal outgrowth |
2. Radioulnar (anteroposterior) axis |
3. Dorsoventral axis |
B. Failure of axis formation/differentiation – hand plate |
1. Radioulnar (anteroposterior axis) |
2. Dorsoventral axis |
C. Failure of hand plate formation/differentiation – unspecified axis |
1. Soft tissue |
2. Skeletal deficiency |
3. Complex |
2. Deformations |
A. Constriction ring sequence |
B. Arthrogryposis |
C. Trigger digits |
D. Not otherwise specified |
3. Dysplasias |
A. Hypertrophy |
1. Macrodactyly |
2. Upper limb |
3. Upper limb and macrodactyly |
B. Tumorous conditions |
Syndactyly Classification
Syndactyly is anatomically subdivided into simple incomplete (Fig. 1a) and complete (Fig. 1b) forms where only the soft tissues are fused, complex forms with adjacent bone fusion like acrosyndactyly (Fig. 1c), and complicated forms where the anatomy is completely subverted (Fig. 1d). Temtamy and McKusick in 1978 proposed a clinical classification of syndactyly, based on the affected digits. They subdivided syndactyly into five groups:
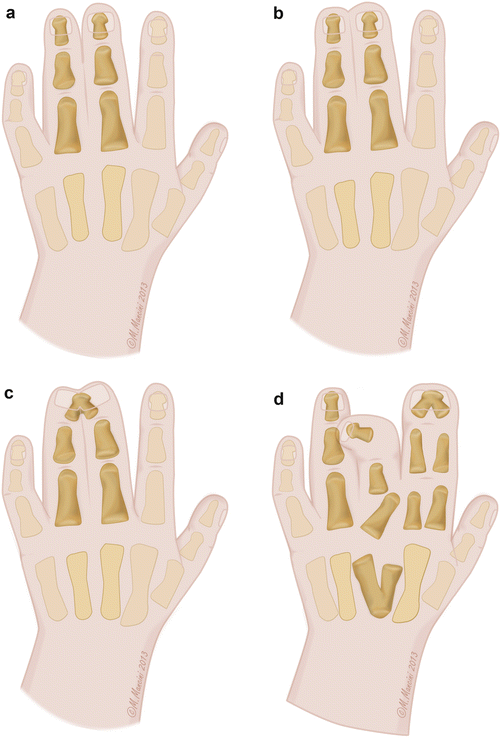
Fig. 1
(a) Simple incomplete syndactyly (Courtesy Marisa Mancini, Medical Illustrator, Modena, Italy). (b) Simple complete syndactyly (Courtesy Marisa Mancini, Medical Illustrator, Modena, Italy). (c) Complex syndactyly (acrosyndactyly) (Courtesy Marisa Mancini, Medical Illustrator, Modena, Italy). (d) Complicated syndactyly (synpolydactyly) (Courtesy Marisa Mancini, Medical Illustrator, Modena, Italy)
Syndactyly type 1 (SD1): the most common type is sporadic or inherited (autosomal dominant with incomplete penetrance), affecting the third web in the hand and/or second web in the foot; it may be incomplete or complete, and most of the times it is bilateral. Inherited forms have been further subdivided into four subgroups depending on the different combinations of hand and foot involvement, one of which (Castilla type) is peculiar as it is characterized by fourth web involvement in the foot.
Syndactyly type 2 (SD2): synpolydactyly (SPD), affecting the third web in the hand and the fourth web in the foot (Fig. 2a, b), it presents with partial or complete duplication within the syndactylous webs. Several subtypes have been described due to the heterogeneity of the spectrum of anomalies.
Syndactyly type 3 (SD3): sporadic or syndromic (oculodentodigital syndrome), affecting fourth web in the hand bilaterally.
Syndactyly type 4 (SD4): rare type, autosomal dominant inherited, presenting with complete syndactyly of all digits in the hand.
Syndactyly type 5 (SD5): rare type, autosomal dominant or X-linked recessive inherited. This type of syndactyly is characterized by metacarpal and metatarsal synostoses and usually affects the fourth web in the hand and third web in the foot.
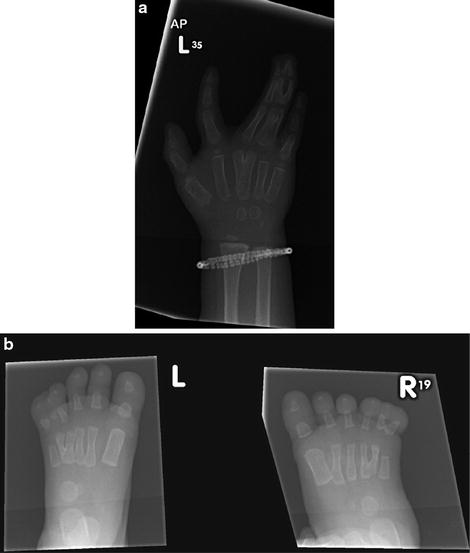
Fig. 2
(a) Radiographic appearance of a hand in a case of synpolydactyly. (b) Radiographic appearance of feet in a case of synpolydactyly
Four additional types of syndactyly have been described since Temtamy and McKusick’s original classification:
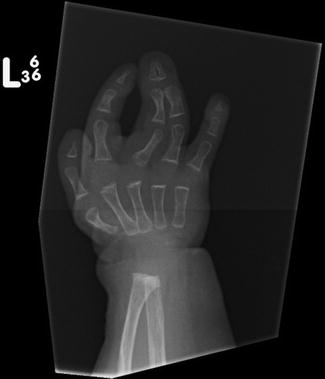
Syndactyly type 6 (SD6): very rare, characterized by unilateral second to fifth finger fusion.
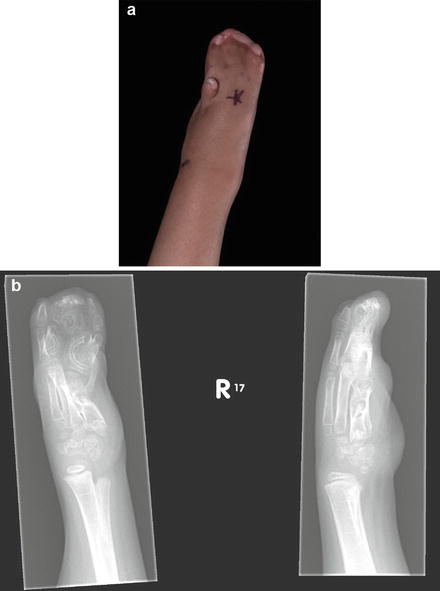
Syndactyly type 7 (SD7): this is the very rare Cenani-Lenz syndrome, autosomal recessive inherited, with complicated anomalies that make the hands and feet appear as indistinct masses of digits (see section “Cenani-Lenz Syndrome”) (Fig. 3a, b).
Syndactyly type 8 (SD8): very rare, fourth and fifth metacarpal synostosis.
Syndactyly type 9 (SD9): mesoaxial synostotic syndactyly (MSSD), described only in two families. Described features include complex third web syndactyly with proximal phalanges size reduction, hypoplastic thumbs and halluces, second and fifth fingers middle phalangeal hypoplasia/aplasia, simple incomplete or complete syndactyly of the toes.
Fig. 3
(a) Hand appearance in a case of Cenani-Lenz syndrome. (b) Radiographic appearance of the hand
Syndromes Associated with Syndactyly
Syndactyly, as part of a syndrome, may present either as a sporadic or a genetically inherited anomaly and may involve the webs in different combinations in the hands and the feet, often bilaterally. The spectrum of anomalies is highly variable, as any subtype of syndactyly may occur, ranging from simple incomplete forms to complex and complicated forms, including those associated with clefting of the hands and feet, polydactyly, and oligodactyly. The associated syndromes include a large number of different clinical entities, and the continuous discovery of new syndromes renders this a fascinating subject of investigation. The most important syndromes associated with syndactyly are listed, with considerations that may specifically affect management and consideration for surgery and anesthesia highlighted:
Amniotic Bands Syndrome (Constriction Rings Syndrome; Congenital Constricting Bands; Amniotic Bands Sequence; ABS; Streeter Anomaly)
[MIM 217100]
Etiology/inheritance: sporadic, with no evidence of a clear genetic origin. There are extrinsic theories of formation, where rupture of the amnion with subsequent constriction of body parts displaced through is causative, and intrinsic theories, related to loss of signaling, possibly due to a vascular problem creating malformations.
Estimated incidence: 1 in 1,200–1 in 15,000 live births.
Findings: ringlike constrictions that may be located anywhere in the body but more frequently in the limbs (Fig. 4), congenital amputation, syndactyly, polydactyly, encephalocele, facial/orbital clefts, eyelid coloboma, cleft lip/palate, ectopic heart, abnormal lung lobation, thoracoschisis, abdominoschisis, gastroschisis, omphalocele, bladder exstrophy, and scoliosis.
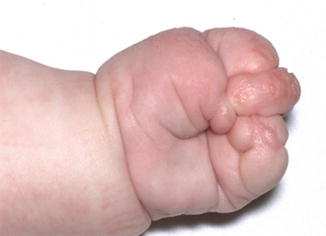
Fig. 4
Acrosyndactyly and amniotic bands
Diagnosis: based on the typical clinical findings at birth. When unilateral, this is often confused with symbrachydactyly, but the presence of multiple rings and the absence of distal nail elements in amputations should clarify this. Prenatal diagnosis is possible in some cases by ultrasound examination (the earliest documented detection at approximately 12 weeks’ gestation).
Apert Syndrome
[MIM 101200]
Etiology: sporadic or autosomal dominant (FGFR2 gene mutation, chromosomal location 10q26.13). Apert syndrome appears to occur more often in children of older fathers.
Estimated incidence: 1 in 160,000–1 in 200,000 live births.
Features: craniosynostosis, corpus callosum agenesis, midfacial hypoplasia (Fig. 5a), pulmonary agenesis, cardiac defects, genitourinary anomalies, esophageal atresia, tracheoesophageal fistula, cervical vertebrae fusion, developmental delay, complex syndactyly (hands and feet), and symphalangism of the interphalangeal joints of all the fingers except the little finger distal interphalangeal (DIP) joint.
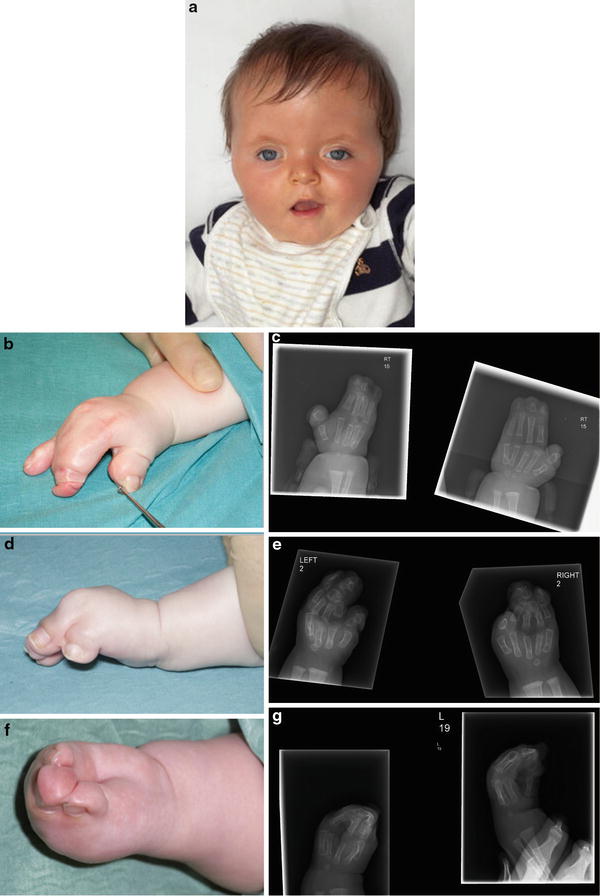
Fig. 5
(a) Facial appearance in Apert syndrome. (b) Type 1 hand in Apert syndrome. (c) Radiographic appearance of type 1 hand. (d) Type 2 hand in Apert syndrome. (e) Radiographic appearance of type 2 hands. (f) Type 3 hand in Apert syndrome. (g) Radiographic appearance of type 3 hand
Their craniofacial anomalies make them prone to airway problems including obstructive sleep apnea, and raised intracranial pressure. The treatment of these entities takes priority over the hands.
Apert’s hand syndactyly is subdivided into three types, according to Upton’s classification (1991):
Type 1 (“spade hand”): the thumb is free but radially deviated due to a delta proximal phalanx, and the palm is flat with complex acrosyndactyly (osseous or cartilaginous) of third web, simple syndactyly of fourth web, and syndactyly of the second web which may be simple or complex (Fig. 5b, c).
Type 2 (“mitten hand”): simple syndactyly of first web with short radially deviated thumb, cupped palm with complex acrosyndactyly of second and third web, and simple complete syndactyly of fourth web (Fig. 5d, e).
Type 3 (“rosebud hand” or “hoof hand”): complete syndactyly of all the digits, with complex (and often complicated) syndactyly of first, second, and third web, and simple syndactyly of fourth web. There is a common nail, prone to paronychial infections, and it may be difficult to distinguish the individual digits within the syndactyly mass, either clinically or radiologically (Fig. 5f, g).
Capitohamate carpal coalition is frequent as is the presence of fourth and fifth metacarpal synostosis, which becomes more evident radiologically with increasing age. Index finger deviation due to an abnormal epiphysis in the proximal phalanx may occur, particularly in type 3 hands.
The foot deformities are similar to those affecting the hands, with multiple tarsal coalitions, radiologically more evident with age. There is multidigit syndactyly with symphalangism, metatarsal synostosis, and occasional polydactyly.
Diagnosis: based on the typical clinical findings at birth of the facial appearance combined with multidigit syndactyly in all limbs. Genetic testing can confirm the diagnosis.
Carpenter Syndrome (Acrocephalopolysyndactyly Type II; ACPS II)
[MIM 201000]
Etiology/inheritance: autosomal recessive (RAB23 gene mutation, chromosomal location 6p11.2).
Features: syndactyly (hands and feet), camptodactyly, brachydactyly (hands and feet), polydactyly (hands and feet), obesity (from childhood), craniosynostosis, hydrocephalus, brachycephaly, mandibular/maxillary hypoplasia and other distinctive facial features (low-set ears, abnormal eye shape, flat nasal bridge), cardiac defects (including dextrocardia in some cases), corneal opacity, genital anomalies in affected males (most frequently cryptorchidism), situs viscerum inversus in some cases, and mild to severe intellectual disability in some cases. Many of the same considerations apply to these children as to the Apert patients, but their hand syndactyly is less severe.
Diagnosis: based on clinical and radiological features and genetic testing.
Cenani-Lenz Syndactyly Syndrome (Cenani-Lenz Syndrome , CLS, CLSS)
[MIM 212780]
Etiology/inheritance: autosomal recessive (LRP4 gene mutation, chromosomal location 11p12-p11.2).
Estimated incidence: unknown. Extremely rare syndrome, with only around 30 cases described in the literature.
Features: bilateral complex syndactyly of the hands and feet with characteristic symmetrical appearance. There is multidigit complex syndactyly with carpal, metacarpal, and phalangeal synostoses; toe syndactyly with tarsal, metatarsal, and phalangeal synostoses; and frequent oligodactyly. Symphalangism may be present. The forearms are also affected, with radial and ulnar hypoplasia, radial head dislocation, radioulnar synostosis, and brachymesomelia. Facial dysmorphism (ptosis, prominent forehead, flat nasal bridge, hypertelorism, downslanting palpebral fissures) has been described in some cases. Renal anomalies (agenesis and/or hypoplasia) have been demonstrated to affect over 50 % of CLS families (Li et al. 2010).
These patients, who have little in terms of muscle and tendon attachments distally, combined with their complex synostoses, may get minimal gain from surgery, and the surgeon should be cautious in his or her prognosis for functional improvement after surgery.
Diagnosis: clinical and radiological features and genetic testing.
Deletion of 2q37 (Monosomy 2q37)
Etiology/inheritance: sporadic (chromosomal disorder, characterized by deletion of chromosome band 2q37).
Estimated incidence: 1 in 10,000 live births.
Features: microcephaly or macrocephaly, typical facial dysmorphism (round cheeks/round face, upslanting palpebral fissures, prominent forehead, midfacial hypoplasia, enophthalmos, sparse scalp hair, sparse and arched eyebrows, depressed nasal bridge, deficient nasal alae, V-shaped nose tip, prominent columella, thin vermillion border of the lips, high-arched palate), fifth finger clinodactyly, small feet and/or hands, hands/feet syndactyly, persistent fetal finger pads, single palmar crease, wide set or distally placed nipples, and supernumerary nipples. Proximal implantation of the thumbs and short toenails has been described in a case. Eczema and hypotonia are frequent findings. Congenital heart malformations and gastrointestinal, genitourinary, and central nervous system malformations have been reported. Seizures have also been described, and behavioral anomalies (a distinct subtype of autism) associated with this disorder have been reported. Albright hereditary osteodystrophy-like phenotype (AHO3) is part of this spectrum, and its features are including developmental delay or intellectual deficit, short stature, tendency towards obesity with age, and brachymetaphalangism.
Diagnosis: based on clinical and radiological features and genetic testing.
Diploid/Triploid Mosaicism (Diploid/Triploid Mixoploidy; 2n/3n Mixoploidy)
Etiology/inheritance: chromosomal disorder (46XX/69XXY), with a mosaic morula (2n/3n) due to partial recovery of a triploid conception (see section “Triploidy Syndrome”). The fetus may not survive.
Features: hands/feet syndactyly, body and/or facial asymmetry, hypotonia, truncal obesity, growth delay, hyperpigmentation or hypopigmentation of the skin, malformed low-set ears, small phallus, micrognathia, and variable intellectual disability (van de Laar et al. 2002; Rittinger et al. 2008).
Diagnosis: clinical features may lead to suspicion. Only genetic testing can confirm the diagnosis (amniocentesis, karyotype of a newborn baby or miscarried/stillborn fetus).
Ectrodactyly-Ectodermal Dysplasia-Cleft Lip/Palate Syndrome (EEC Syndrome)
Due to genetic variability different forms of this disorder are known:
Ectrodactyly-Ectodermal Dysplasia-cleft lip/palate syndrome 1 (EEC1)
[MIM 129900]
Ectrodactyly-Ectodermal Dysplasia-Cleft Lip/Palate syndrome 3 (EEC3)
[MIM 604292]
Etiology/inheritance:
EEC1: autosomal dominant with incomplete penetrance (linked to chromosomal location 7q11.2-q21.3)
EEC3: autosomal dominant with incomplete penetrance (TP63 gene mutation, chromosomal location 3q28)
Features: EEC is characterized by high variability, and no sign appears to be obligatory for the diagnosis (Fryns et al. 1990). Described features are syndactyly of the hands/feet, ectrodactyly of the hands/feet (Fig. 6a–d), maxillary and mild malar hypoplasia, malformed/small ears, hearing loss, flat nasal tip, cleft lip, cleft palate, absence of Stensen duct, xerostomia, microdontia, teeth anomalies (hypodontia, partial anodontia, enamel hypoplasia), dental caries, lacrimal duct abnormalities, blepharophimosis, blue irides, blepharitis, dacryocystitis, photophobia, sparse eyebrows, sparse eyelashes, sparse and thin scalp hair, sparse pubic and axillary hair, slow hair growth, light colored hair, fair and thin skin, periorbital hyperpigmentation, dystrophic/pitted/thin nails, hypoplastic nipples, micropenis (male), cryptorchidism (male), transverse vaginal septum (female), renal agenesis/dysplasia, hydronephrosis, duplicated collecting system, megaureter, ureterocele, vesicoureteral reflux, bladder diverticula, holoprosencephaly, mental retardation in some cases, growth hormone deficiency, hypogonadotropic hypogonadism, and central diabetes insipidus.
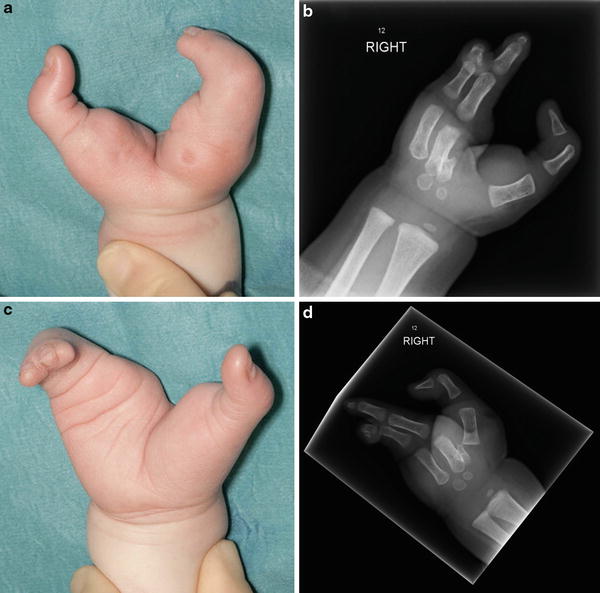
Fig. 6
(a) Clinical appearance of the right hand ectrodactyly in a case of EEC syndrome. (b) Radiographic appearance of the right hand. (c) Clinical appearance of the left hand ectrodactyly. (d) Radiographic appearance of the left hand
Diagnosis: based on clinical and radiological features (high variability) and genetic testing.
Filippi Syndrome (Syndactyly Type 1 with Microcephaly and Mental Retardation; Scott Craniodigital Syndrome with Mental Retardation)
[MIM 272440]
Etiology/inheritance: autosomal recessive (unknown genetic cause).
Features: hands/feet syndactyly, polydactyly, microcephaly, growth delay, low weight, distinctive facial features (micrognathia, short philtrum, broad forehead, high frontal hairline, hairy forehead, prominent columella, broad nasal bridge, hypoplastic nasal alae), and variable mental retardation/intellectual disability. Other reported features included phalangeal and metacarpal hypoplasia, cryptorchidism (male), teeth and hair abnormalities, hypertonia of the limbs, central hypotonia, carpal synostosis, radial head dysplasia and elbow dislocation, anteverted ears with hyperconvoluted helix, thin lips, generalized hirsutism, ventricular septal defect of the heart, Chiari type I brain malformation, cerebellar atrophy, diffuse enlargement of subarachnoid spaces and lateral ventricles, megacisterna magna, and arachnoidal cyst. Dystonic movements, dystonic tongue protrusion, and seizures have also been reported.
Diagnosis: based on typical clinical and radiological features.
Fraser Syndrome (Cryptophthalmos-Syndactyly Syndrome)
[MIM 219000]
Etiology/inheritance: autosomal recessive (homozygous or compound heterozygous mutation in FRAS1 located on chromosome 4q21, FREM2 on chromosome 13q13, or GRIP1 on chromosome 12q14).
Features: Fraser syndrome is a heterogeneous disorder, with several different features reported in different individuals. Reported features have been cryptophthalmos (this finding is not essential, as affected individuals may not have it) (Koenig and Spranger 1986; Thomas et al. 1986), hypertelorism (Fig. 7a), blindness, absent or malformed lacrimal ducts, middle end external ear malformations, hearing loss or congenital deafness, hypoplastic notched nares, broad and low nasal bridge, midline nasal cleavage, microcephaly, unusual hairline (hair growth on temples), high palate, cleft lip/palate, laryngeal stenosis or atresia, renal hypoplasia or agenesis, umbilical abnormalities, small penis (male), hypospadias (male), cryptorchidism (male), bicornuate uterus (female), vaginal atresia (female), clitoral enlargement (female), imperforate anus or anal stenosis, pubic symphysis diastasis, meningomyelocele, encephalocele, and mental retardation in some cases. Rate of prenatal death is 25 %, while 20 % of affected individuals die before age one.
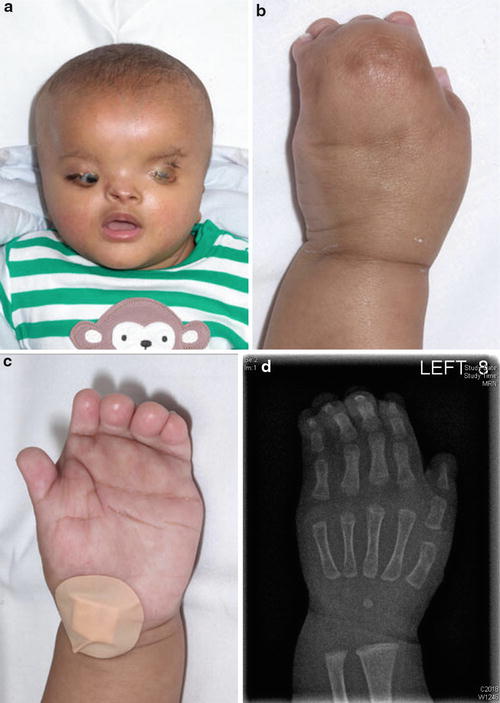
Fig. 7
(a) Facial appearance in Fraser syndrome. (b) Clinical appearance of the left hand (dorsal view) (c) Clinical appearance of the left hand (palmar view). (d) Radiographic appearance of the left hand
Diagnosis: diagnostic criteria were established by van Haelst and Scambler in 2007:
Major criteria: syndactyly (Fig. 7b, c, d), cryptophthalmos, urinary tract abnormalities, ambiguous genitalia, laryngeal and tracheal anomalies, and positive family history
Minor criteria: anorectal defects, dysplastic ears, skull ossification defects, umbilical abnormalities, and nasal anomalies
All the other malformations and anomalies were considered uncommon. Diagnosis can be made if either 3 major, or 2 major and 2 minor, or 1 major and 3 minor criteria are present in the same individual.
Goltz Syndrome (Focal Dermal Hypoplasia; FDH; FODH; DHOF; Goltz-Gorlin Syndrome)
[MIM 305600]
Etiology/inheritance: sporadic (95 % of cases), or X-linked dominant (PORCN gene mutation, chromosomal location Xp11.23) with in utero lethality in males. Affected males are all results of new mutation.
Features: poikiloderma with focal dermal hypoplasia, atrophy and linear pigmentation of the skin with fatty tissue herniation through the dermal defects, multiple papillomata of the mucous membranes (external and internal) and skin, hands/feet syndactyly (Fig. 8), polydactyly, oligodactyly, brachydactyly, hypoplastic phalanges/metacarpals/metatarsals, and split hand. The “lobster-claw hand” (i.e., combination of split hand, ray absence, and syndactyly) is a typical feature of FDH, and similar deformities have been reported also affecting the foot. Longitudinal striation of the long bones (osteopathia striata) has been reported as a common finding. Mental retardation is frequent.