Surgical Care of Conditions Presenting in the Newborn
Gary E. Hartman
Michael J. Boyajian
Sukgi S. Choi
Martin R. Eichelberger
Kurt D. Newman
David M. Powell
Successful management of newborns with surgical conditions requires close cooperation among neonatologist, surgeon, anesthesiologist, and radiologist. The fragile nature and limited reserve of these infants are superimposed on the stress of the surgical condition and its operative correction. In addition to the physiologic concerns related to the surgical condition, special attention must be directed toward (a) temperature regulation, (b) fluid, blood, and glucose administration, and (c) monitoring of respiratory and cardiovascular performance.
Venous and arterial access for fluid administration and monitoring may be challenging. Peripheral venous access with a 22- or 24-gauge catheter provides adequate access for the most vigorous fluid resuscitation. Central venous access may be necessary in the unstable newborn or if peripheral access is unsuccessful. Umbilical artery catheterization provides vascular access and arterial monitoring and is a well-established procedure in even the smallest prematures. The umbilical catheter may be maintained in most operative procedures. Peripheral arterial access may be obtained using radial, ulnar, or posterior tibial puncture or cutdown.
Fluid requirements are usually significantly greater than maintenance, especially in situations with intestinal obstruction, peritonitis, or gastroschisis. Before the operation there may be extraordinary losses from the gastrointestinal (GI) tract or inflamed peritoneum. These losses continue in the postoperative period and are superimposed on the sodium and water retention associated with the stress response. The endocrine, metabolic, and cytokine response to operative stress, which has been well documented in adults, has been confirmed in neonates (1,2 and 3). Postoperatively, decreased urine volume may result from the surge in antidiuretic hormone, intravascular volume depletion, or both, limiting the utility of urine volume alone as a monitor of adequacy of fluid replacement. Additional assessments including skin temperature, quality of peripheral pulses, serial measurements of weight, he-matocrit, and serum electrolytes and osmolality supplement urine volume as indicators of adequate volume replacement (4).
Temperature maintenance is a critical concern for all newborns, but in particular for those undergoing diagnostic studies and operative procedures in radiology and operating suites. A variety of means of temperature support may be utilized, including warming the environment, heat lamps, heating blankets, scalp and extremity wrapping, heated intravenous fluids and blood products, and heated inhaled anesthetic agents and surgical irrigating fluids. Temperature must be constantly monitored, and every effort made to minimize exposure to cold stress. Hypothermia is a potentially lethal condition, and the importance of temperature support cannot be overemphasized (5).
Anesthetic and postoperative pain management can minimize the magnitude of the stress response and accelerate the neonate’s return to normal homeostasis in terms of cortisol, catecholamine, and insulin modulation (6). The expertise of all the specialists involved may be the advantage necessary to achieve survival and mandates close collaboration.
LESIONS OF THE HEAD AND NECK
Congenital abnormalities of the head and neck occur commonly in the newborn. Because of the short, fat neck of the baby, some of these lesions are not immediately apparent, and the examiner must be alert to their possibility to detect them.
Cleft Lip
Clefts of the lip or palate occur in approximately one of every 600 to 700 Caucasian newborns. The frequency is doubled in Asians and halved in African Americans. Cleft lip occurs somewhat more often in male patients and on the left side. The defect probably results from lack of the mesodermal reinforcement of the junction of the nasome dial and lateral facial processes that normally takes place in the sixth to seventh week of gestation. Multiple genetic influences seem to be more important than environmental factors. The cleft deformity ranges from minor notching to complete separation of the entire lip and nasal floor (Fig. 44-1). The defect may involve the lip, the lip and palate, or only the palate, and it may be unilateral or bilateral. Median cleft lip is rare and is usually associated with hypotelorism, microcephaly, and early death.
Airway obstruction is not typically a consequence of isolated cleft lip or palate. Initial care focuses on feeding the infant and counseling the parents. Swallowing and airway protection should be normal, but the negative pressure of the normal suck is vented through the cleft, resulting in inadequate inflow. Fatigue during feeding is common and may mimic satiation. Although suckling is not altogether discouraged, a baby with a complete cleft lip or any degree of cleft palate should be expected to suffer mechanical feeding difficulty. The solution may be to rely on an enlarged nipple aperture, a compressible bottle, or a syringe feeder. With the use of a positive-pressure delivery system, the feeding schedule should be normal.
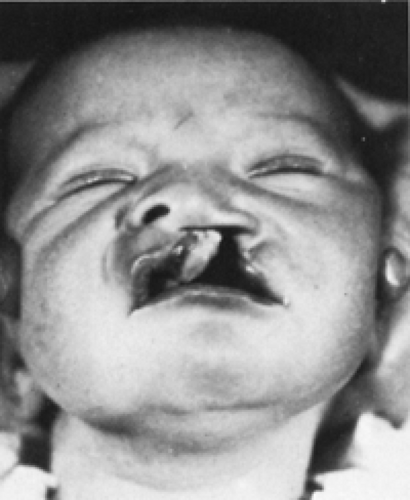 Figure 44-1 Complete congenital cleft of the lip with associated cleft of the palate that extends forward through the alveolar ridge. |
Lip closure is usually carried out at around 3 months of age. The major goals are muscle continuity, balanced lip height, the normal Cupid’s-bow lip shape, a smooth and pout-free lip margin, a good nasal sill, adequate sulcus lining, and a minimal, well-placed scar. The wide, complete unilateral and the complete bilateral clefts present greater challenges. Preliminary lip adhesion for the unilateral case or presurgical orthodontics can improve anatomic associations and facilitate the definitive surgery. Residual nasal deformity is often a stubborn problem and may require secondary surgery.
Cleft Palate
The embryologic palatal shelves initially hang vertically and then rise to meet and fuse from front to back between weeks 7 and 12 of gestation. Interference with this process may result in complete, incomplete, or submucous cleft of the palate. Initial care is discussed in the section on cleft lip.
The major significance of this defect is the effect on speech. Normal modulation of speech requires reliable, dynamic palatal separation of the mouth from the nose. This requires a palate of adequate length, suppleness, and muscle power. Velopharyngeal incompetence or incomplete nasal closure results in hypernasal speech and significant communication disability.
Chronic or recurrent effusion and infection in an otherwise normal ear is common in the child with a cleft palate because eustachian tube function is compromised. This child usually needs myringotomies and ventilation tubes.
Early surgery seems to have a negative effect on facial growth, but the trend is toward closure during infancy because of the improved speech results. Most American surgeons choose 9 to 12 months of age as optimal timing for a single-stage closure.
Palatal closure is accomplished with local soft tissue. Mucoperiosteal flaps are mobilized and closed in the midline, with oral and nasal lining, effecting muscle apposition and retroposition. No bone reconstruction is involved. The goal is normal speech, and this is achieved in approximately 85% of patients. A second operation produces good results for almost all the remaining infants.
An essential concept in the treatment of these children is a multidisciplinary approach. The patient should be followed through adolescence by a team consisting of a plastic surgeon, otolaryngologist, audiologist, pedodontist, orthodontist, speech pathologist, geneticist, pediatrician, and social worker.
Pierre-Robin Sequence
The Pierre-Robin sequence is characterized by retrognathia or microgenia (i.e., small or recessed jaw or chin), glossoptosis, airway obstruction, and cleft palate. The lack of forward support of the tongue allows it to fall back and
compromise the airway. The basic defect may result from intrauterine restriction of mandibular growth.
compromise the airway. The basic defect may result from intrauterine restriction of mandibular growth.
Intensive monitoring, including a home apnea monitor, is necessary for many patients. The airway can usually be maintained by conservative measures. Prone positioning allows the tongue to fall forward. An appropriately apertured board may facilitate this positioning, and a nasal airway may be useful. Early gavage feedings may obviate hazardous oral feedings. A lip-tongue adhesion may be performed in more difficult cases, but its effectiveness varies. Tracheostomy should be avoided, if possible, but it is sometimes the only safe choice. Management should be as conservative as the clinical situation permits. The airway problem is typically self-limited, resolving as the child grows.
MASSES IN THE NECK
Masses in the neck are common in children and may be congenital, infectious, or neoplastic. Thyroglossal duct remnants, branchial apparatus anomalies, and lymphangiomas (i.e., cystic hygromas) are the most common congenital pediatric lesions in the neck.
Thyroglossal Duct Remnants
Thyroid tissue left behind in an abnormal location during normal developmental descent can result in a thyroglossal duct cyst, which presents in the midline of the neck. Infection may lead to a cutaneous salivary fistula. After appropriate therapy for infection, treatment consists of resection of the cyst, the central portion of the hyoid bone, and dissection of the tract up to its origin at the foramen cecum (Sistrunk operation) (7,8 and 9).
Branchial Cleft Anomalies
Preauricular Tabs and Sinuses
These lesions are not true branchial cleft remnants because they originate from an abnormal formation of the ear rather than a branchial cleft component. The preauricular sinus almost always ends blindly, and excision is indicated to prevent recurrent infection in later years.
Cervical Fistulas
Fistulas that originate from the first branchial arch present in the neck just below the ear and communicate with the external auditory canal. A fistula originating from the second branchial arch is the most common branchial cleft remnant. The fistula usually extends from the skin of the lower neck upward along the sternocleidomastoid muscle and then passes inward between the internal and external carotid arteries to attach to the posterolateral pharynx just below the tonsillar fossa (Fig. 44-2). The presenting complaint is usually related to persistent or intermittent drainage onto the neck. Complete surgical extirpation is necessary for cure. A large incision in the neck can be avoided by the use of two or three small, neat, transverse stair-step incisions.
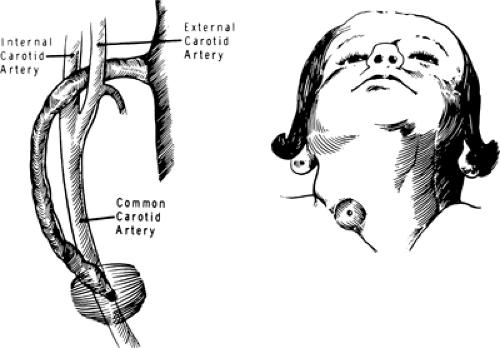 Figure 44-2 Brachial cleft cyst, which presents low in the neck as a mass or an opening in the skin, extends upward and laterally in the neck, and passes between the branches of the carotid artery to connect with the pharynx below the tonsillar facia. From Nardi GL, Zuidema GD. Surgery, 3rd ed. Boston: Little, Brown, 1972, with permission. |
Branchial Cyst
Approximately 10% of persistent branchial deformities are cystic. These invariably arise low in the anterior triangle of the neck and present as smooth cysts anterior to the sternocleidomastoid muscle. Dissection with excision is curative.
Cervical Cutaneous Tabs
Occasionally, a baby presents with a cutaneous tab in the skin of the anterior aspect of the neck. A small, irregular mass of cartilage may be contained in the skin tab. The cartilage is never associated with a fistula, and removal of this small appendage is not urgent.
Cystic Hygroma
Cystic hygroma (i.e., lymphangioma) is a congenital deformity arising from abnormal development of lymphatic channels (Fig. 44-3) (12). About 80% of these watery cysts occur in the neck, and most are located posterior to the sternocleidomastoid muscle. Other sites of occurrence are the groin, the axilla, and the mediastinum. The term “hygroma” suggests the watery fluid contained in the
endothelium-lined spaces. The cyst may be unilocular, but more often there are numerous cysts of various sizes that permeate the surrounding structures and distort the local anatomy. Supporting connective tissue often shows extensive lymphocytic infiltration. Except in the case of a single large cyst, no definite cleavage plane is found between hygroma and normal tissue.
endothelium-lined spaces. The cyst may be unilocular, but more often there are numerous cysts of various sizes that permeate the surrounding structures and distort the local anatomy. Supporting connective tissue often shows extensive lymphocytic infiltration. Except in the case of a single large cyst, no definite cleavage plane is found between hygroma and normal tissue.
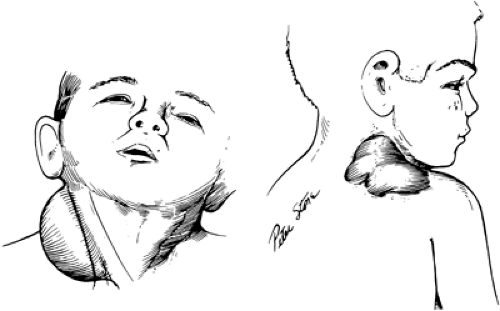 Figure 44-3 The typical cystic hygroma occurs in the lateral neck. The mass may extend into the scapular, axillary, or thoracic compartments, or the hygroma may present separately in any of these locations. Although depicted here as a single cyst, the hygroma is often a multiloculated, ill-defined mass. From Nardi GL, Zuidema GD. Surgery 3rd ed. Boston: Little, Brown, 1972, with permission. |
The lesion is usually evident at birth. Occasionally, the mass occupies the entire submandibular region, distorting the subglottic area and compromising the airway. A supraclavicular mass may become prominent with the Valsalva maneuver. This form of cystic hygroma is usually associated with a mediastinal component. Some cystic hygromas contain nests of poorly supported vascular channels that are prone to bleeding and may produce sudden enlargement and discoloration of the lesion.
Symptoms are related entirely to the location and size of the mass. Disfigurement is often severe. Infection in the mass may lead to dangerous regional cellulitis, but after the infection subsides, the resultant intracystic fibrosis and scar may significantly reduce the size of the tumor mass. Prenatal ultrasonography has been used to diagnose cystic hygroma (13). This modality has demonstrated a hidden mortality with a high incidence of associated anomalies, including abnormal karyotypes and hydrops fetalis, when lymphangiomas are detected before 30 weeks of gestation.
Repeated aspiration of the cyst with injection of sclerosing agents is not recommended because any surgical excision that is subsequently required is rendered significantly more difficult by the sclerosing procedure. Elective surgical excision between 4 and 12 months of age is indicated for asymptomatic patients. Airway compression or recurrent infections may necessitate removal at an earlier age (14). Total excision is often impractical because of the extent of the hygroma and its proximity to vital structures. Important nerves and vascular structures must not be sacrificed in an attempt to achieve total excision of this benign lesion; multiple excisions of the residual hygroma are preferable. Postoperative wound drainage using closed-suction drains may reduce recurrence.
UPPER AIRWAY OBSTRUCTION CAUSING RESPIRATORY DISTRESS
A neonate is essentially an obligate nasal breather for the first several months of life. (Recently this belief has been challenged, and some investigators have proposed that a neonate is a preferential nasal breather [15].) Therefore, during the first few months of an infant’s life, any nasal condition causing obstruction can cause respiratory difficulties. Typically a neonate with nasal obstruction presents with cyclic cyanosis (See Color Plate).
A newborn’s epiglottis is softer and bulkier than that of older children and adults and is often tubular in shape. Excessive and redundant mucosa over the epiglottis and arytenoids can contribute to inspiratory stridor. The larynx in a newborn is a well-developed organ; however, it is one-third the size of an adult larynx. The subglottis of a neonate measures approximately 4.5 mm in diameter. Because of the smaller dimension, 1 mm of circumferential edema in the subglottis can reduce the cross-sectional area of a neonate’s airway by more than 60% (16). The trachea and bronchi are also smaller in dimensions and are shorter than those in an adult. In addition to these anatomic dif-ferences, the airway structures in a neonate are more pliable, and its mucosa more reactive. Therefore, conditions that affect the airway such as epiglottitis and croup can have proportionately greater effect on a small infant’s airway.
Evaluation of airway obstruction begins with a careful history regarding the characteristics of the stridor or other upper airway noises, voice quality, severity of airway symptoms, feeding difficulties, previous intubation history or other manipulation of the airway, other associated anomalies, and symptoms of gastroesophageal reflux or aspiration. The great majority of airway abnormalities causing obstructive symptoms can be identified from the history alone (17). History is supplemented by physical examination and other evaluation modalities. High-kilovolt anterior-posterior and lateral soft tissue film of the neck can help in evaluation of the areas from nasal cavity to subglottis. Posterior-anterior and lateral chest x-rays can help to delineate tracheal and bronchial pathology. Other studies such as computed tomography (CT) scans and magnetic resonance imaging (MRI) may be needed to evaluate airway obstruction involving the nasal cavity, nasopharynx, and trachea.
Flexible nasopharyngoscopy and laryngoscopy can be performed at the bedside with monitoring. Areas from nasal vestibule to the vocal cords can be evaluated for functional and anatomic abnormalities. Flexible bronchoscopy with sedation is often suboptimal for evaluation of the subglottic, tracheal, and bronchial airways and is inferior to rigid endoscopy. Rigid endoscopy is usually performed in the operating room under general anesthesia but with
spontaneous ventilation (no paralysis). This type of anesthesia enables the surgeon to evaluate the dynamics of the airway and any anatomic abnormalities.
spontaneous ventilation (no paralysis). This type of anesthesia enables the surgeon to evaluate the dynamics of the airway and any anatomic abnormalities.
Nasal Obstruction
Nasal Pyriform Aperture Stenosis
Congenital nasal pyriform aperture stenosis (anterior nasal stenosis) is secondary to the overgrowth of the nasal process of the maxilla (18). In this condition, the entrance into the nasal cavity known as the pyriform aperture is reduced to a slit-like opening. Because the pyriform aperture is the narrowest part of the nasal airway, even small changes in the cross-sectional area at this level can result in significant increase in nasal airway resistance and airway obstruction. Clinically, nasal obstruction causes apneic episodes and cyclic cyanosis, which is relieved by crying. Physical examination shows a bony, shelf-like projection of the posterior portion of the vestibule that almost completely obstructs the nasal cavity. The CT scan is the study of choice and helps to confirm the diagnosis and to define the anatomy of the nasal cavity and posterior choanae. Association of anterior nasal stenosis with midfacial dyso-stosis and endocrine and central nervous system abnormalities has been reported (19). Therefore, a genetic consult should be considered.
The infant is initially managed with a McGovern nipple or an oral airway with close monitoring. Gavage feeding may be necessary. If the infant is doing well, then discharge to home with an apnea monitor and a plan for close follow-up evaluations can be considered. If the infant does not tolerate an oral airway or continues to have significant nasal obstruction despite conservative management, then surgical intervention is necessary before discharge from the hospital. Sublabial approach with the use of microinstruments to remove portions of the nasal process of the maxilla is recommended (18). When the obstruction is mucosal rather than bony in nature, the diagnosis is likely to be stuffy nose syndrome, and it requires symptomatic medical treatment only.
Deviated Nasal Septum
Careful intranasal examination shows some septal deformities in as many as 70% of newborns, which may be secondary to intrauterine or birth trauma (20,21). Significant deviation of nasal septum secondary to traumatic delivery or the use of forceps is seen in approximately 1% of neonates. Physical examination shows tilted columella, deviation of the nasal septum and asymmetry of the nasal alae. Radiographic studies are not helpful. Closed reduction of the nasal septum by the use of gentle traction should be carried out during the first week of life. Careful examination of the nasal cavities to detect septal hematoma is essential. If this is present, a septal hematoma must be evacuated emergently because hematoma can lead to abscess formation and saddle nose deformity. Parents should be counseled that nasal growth disturbances can occur following trauma and that further nasal surgery may be necessary when the child is older.
Choanal Atresia
The simplest and the most widely held theory regarding pathogenesis of choanal atresia is that it results from the persistence of bucconasal membranes that normally involute during the seventh week of gestation. The incidence of choanal atresia is approximately one in 5,000 to 8,000 live births. Unilateral choanal atresia is twice as frequent as bilateral choanal atresia. Ninety percent of the atresia is bony; 10% is membranous. The female-to-male ratio is reported to be 2:1. In approximately 50% of the patients with choanal atresia, other associated anomalies are seen. The most common anomaly associated with choanal atresia is the CHARGE association (coloboma, heart disease, atresia of the choana, retarded growth and development, genitourinary anomalies, ear anomalies and/or deafness) (22). For these reasons, a genetic consultation may be indicated for patients diagnosed with choanal atresia.
In cases of unilateral atresia, unilateral mucoid nasal rhinorrhea may be seen; however, significant respiratory distress is usually absent. In bilateral choanal atresia, characteristic cyclic respiratory obstruction is seen. Nasal ob-struction leads to increasing respiratory effort and distress until the child cries and the nasal obstruction is bypassed temporarily. Diagnosis is established by the inability to pass a 6-Fr suction catheter through the nostril beyond 3 to 4 cm into the nasopharynx. The atretic plate can also be visualized using a fiberoptic nasopharyngoscope. The best method of delineating the atresia is by CT scan.
A neonate with bilateral choanal atresia is initially managed with oral airway or McGovern nipple and gavage feedings. Tracheotomy is rarely needed. Choanal atresia can be addressed by transnasal, transpalatal or endoscopic approaches (23,24). Emergent surgical correction for bilateral choanal atresia is seldom required and is reserved for those infants that can not be managed conservatively. Under these circumstances, a transnasal repair can be performed with a staged definitive transpalatal repair when the child is older. In a neonate who is easily managed by conservative means alone, surgical correction can be de-ferred for 1 to 2 years. Restenosis requiring revision surgery is not uncommon.
Congenital Midline Nasal Masses
Encephaloceles and gliomas arise from faulty closure of the foramen caecum at the third week of fetal development (25). Gliomas are locally aggressive lesions that can cause symptoms by enlargement and pressure effects on the surrounding structures. Approximately 30% of gliomas are intranasal and thus can cause nasal obstruction and septal deviation. Fifteen percent of gliomas have a fibrous connection to the dura. Basal encephaloceles herniate through a defect in the cribriform plate and usually present as intranasal masses and cause nasal obstruction.
Congenital midline nasal masses are best evaluated by a combination of CT scan and MRI. Depending on whether there is a connection to the dura, gliomas may require a combined approach by the neurosurgery and otolaryngology services. Encephaloceles always have an intracranial connection and thus require an intracranial exploration. Early surgical intervention is advised to decrease the risk of meningitis and further enlargement of the mass with resulting cosmetic deformity.
Oropharyngeal Obstruction
Glossoptosis
Pierre-Robin sequence (PRS) consists of micrognathia, glossoptosis, and U-shaped cleft palate; it represents the best-known cause of glossoptosis. Pierre-Robin sequence results secondary to arrest in development of mandible at the seventh to the 11th week of gestation, which then leads to high position of tongue in the oral cavity and prevention of fusion of palatal shelves (26). It may present as an isolated anomaly or as a part of a syndrome such as Stickler’s syndrome (see Chapter 38). Genetic consultation is advised.
Airway obstruction in neonates with PRS occurs because of the micrognathia, which causes posterior displacement of the tongue base. Obstruction is more severe in supine position and worsens during sleep or induction of anesthesia. Airway obstruction can be managed by positioning, placement of a nasopharyngeal airway, intubation, and tracheotomy (27). Most patients without other significant airway or neurologic deficits can be managed conservatively (28). For neonates with severe airway symptoms who do not respond to positioning or nasopharyngeal airway, a tracheotomy is required. Once it is performed, the tracheotomy tube remains in place until the cleft palate repair is completed.
Macroglossia
Macroglossia is seen in neonates with Beckwith-Wiedemann syndrome (BWS). Additionally, patients with BWS may have omphalocele, adrenal cytomegaly, and visceromegaly (29). BWS occurs secondary to sporadic mutation and has a mortality rate of over 20%. The airway obstruction seen in these patients is secondary to macroglossia. Initial management consists of tracheotomy, followed by a tongue reduction procedure at a later date.
True macroglossia and relative macroglossia (a small oral cavity) can also be seen in neonates with Down syndrome; however, the size of the tongue rarely necessitates surgical intervention.
Laryngeal Obstruction
Laryngomalacia
Laryngomalacia is the most common cause of stridor in infants (30). Laryngomalacia describes the collapse of supraglottic structures on inspiration. The pathophysiology of laryngomalacia is unknown. Usually, it is a self-limited condition with mild symptoms that resolve by age 18 to 24 months.
Infants with laryngomalacia present with variable in-spiratory stridor that worsens with crying, feedin, and upper respiratory infections and improves with prone position. A small number of infants have more severe symptoms of airway obstruction consisting of retraction, feeding difficulties, failure to thrive, and cyanosis. Diagnosis is confirmed by flexible laryngoscopy, which shows inward collapse of the epiglottis, aryepiglottic folds, and the mucosa over the arytenoids during inspiration. In fewer than 5% of the infants, surgery is indicated to relieve severe obstruction and prevent pulmonary and cardiac complications (31). Surgery consists of trimming the supraglottic tissue with carbon dioxide laser or by sharp dissection (32). A tracheotomy is an alternative to supraglottoplasty (epiglottoplasty).
Vocal Cord Paralysis
Vocal cord paralysis accounts for approximately 10% of all congenital laryngeal anomalies (30). Unilateral paralysis does not cause significant airway symptoms; however, the infant may have a hoarse and breathy cry. Surgical intervention is usually not required. An infant with bilateral vocal cord paralysis (BVCP) presents with normal voice and cry, but with an inspiratory stridor. Central nervous system anomalies, in particular Arnold-Chiari malformation, are often associated with BVCP (30,33). The diagnosis of BVCP is made by flexible laryngoscopy. Radiographs of the airway and chest and, if indicated, CT scan of the brain should be obtained.
In BVCP associated with Arnold-Chiari malformation, neurosurgical decompression will result in resolution of BVCP. In most infants with idiopathic BVCP, tracheotomy is required to establish an airway. During tracheotomy, the entire airway should be inspected to exclude any additional airway anomalies. Definitive vocal cord lateralizing procedures can be done at a later date, if spontaneous resolution of the paralysis does not occur (34).
Laryngeal Atresia/Web
During embryogenesis, epithelium temporarily obliterates the laryngeal lumen. This epithelial plug is then resorbed during the seventh to eighth week of gestation (35). Failure of this resorption process can result in laryngeal web, subglottic stenosis (SGS), and laryngeal atresia. Laryngeal atresia is a rare and life-threatening condition that must be recognized in the delivery room and must be followed by an emergent placement of tracheotomy tube if the infant is to survive.
The most common laryngeal web is seen at the glottic level and affects the vocal cords (36). The web may be thick, causing severe obstruction, or thin and mem-branous. Infants with laryngeal web present with abnormal cry, stridor, and respiratory distress. Laryngeal web can
cause cyanosis or unexplained airway obstruction at birth, which requires intubation or a tracheotomy. Laryngeal web is often associated with SGS. Diagnosis is made by airway films and rigid endoscopy. Definitive treatment options to allow decannulation include dilation, endoscopic microsurgical or laser division of the web, and an open repair (36).
cause cyanosis or unexplained airway obstruction at birth, which requires intubation or a tracheotomy. Laryngeal web is often associated with SGS. Diagnosis is made by airway films and rigid endoscopy. Definitive treatment options to allow decannulation include dilation, endoscopic microsurgical or laser division of the web, and an open repair (36).
Posterior Laryngeal Cleft
Posterior laryngeal cleft is often associated with other congenital anomalies such as esophageal atresia, tracheo-esophageal fistula, and tracheal and bronchial stenosis. Posterior laryngeal cleft is difficult to diagnose, particularly if the cleft is limited to the interarytenoid region. Posterior laryngeal cleft has been classified into four types, with type 1 being a mild interarytenoid cleft above the level of the vocal cords and type 4 representing a complete laryngo-tra cheoesophageal cleft (37).
Presenting symptoms depend on the extent of the cleft. Airway obstruction is not a prominent feature. Rather, aspiration, choking, cyanosis, and feeding difficulties are seen commonly. Stridor and voice abnormalities can be seen but are not common. Diagnosis is made by careful inspection of the posterior glottis during endoscopy. Laryngeal cleft can be repaired endoscopically or by an external approach using laryngofissure or lateral pharyngotomy.
Subglottic Stenosis
SGS is defined as narrowing of the airway at the level of the cricoid to less than 4 mm in a full-term infant and 3 mm in a premature infant. It is considered to be congenital if there has not been any previous airway manipulation. The stenosis may be cartilaginous or membranous. Many of the infants with congenital SGS may have only mild symptoms and go undiagnosed, whereas others may get intubated and thus diagnosed as having acquired SGS. Thus, the true incidence of congenital SGS is unknown. Acquired SGS is most commonly secondary to prolonged endotracheal intubation. One percent to 8% of infants who require prolonged intubation may acquire SGS (38). The injury to the subglottis is related to the duration of intubation, the size of the endotracheal tube (ETT), the degree of ETT motion, and the number of reintubations.
Infants with SGS can present with stridor, respiratory distress, and croupy barking cough. Others present with recurrent or prolonged croup and inability to be extubated after intubation, or obstructive apnea. Airway radiographs can show narrowing of the subglottic airway; however, the definitive diagnosis is made on rigid endoscopy. For infants who have severe airway obstruction and fail repeated attempts at extubation, a tracheotomy or anterior cricoid split (ACS) can be considered. An ACS can be performed only in infants without other underlying airway abnormalities that contribute to the airway obstruction and who have good pulmonary reserve (39). Any need for ventilatory support, oxygen requirement over 30%, congestive heart failure, and respiratory infection are contraindications to ACS.
Anterior cricoid split involves opening of the upper two tracheal rings, cricoid cartilage, and lower half of the thyroid cartilage in the anterior midline and presumably works by decompressing the airway. Postoperative intubation for 7 to 10 days and meticulous care to avoid ETT plugging and accidental extubation are imperative. Systemic corticosteroid is administered beginning 24 hours preextubation and continues for 72 hours postextubation. If ACS is successful, a tracheotomy can be avoided. Patients who fail ACS can undergo revision ACS or a tracheotomy followed by a formal laryngotracheal reconstruction at a later date.
Subglottic Hemangioma
Hemangiomas represent malformation of vasoformative tissue. Hemangioma in the airway usually occurs in the submucosa of the subglottic region; it increases in size over 6 to 18 months, followed by involution (40). Hemangioma may extend beyond the airway into the mediastinum. Approximately 50% of infants with airway hemangioma have cutaneous hemangioma in the head and neck region.
Infants with subglottic hemangioma present with inspiratory stridor, hoarseness, barky cough, and airway obstruction. Airway film shows soft tissue swelling in the subglottis, which is often asymmetric. Endoscopy shows a lesion that is localized to the posterior subglottis and is soft and compressible. Biopsy is usually not necessary unless diagnosis is in question, and the endoscopist should be prepared to manage possible hemorrhage into the airway. Treatment of subglottic hemangioma depends on the degree of airway obstruction and the extent of airway involved by the hemangioma. Treatment options include tracheotomy followed by expectant waiting for involution of the hemangioma, corticosteroids, and laser excision. More recently, interferon-a2a administration has been introduced as a treatment for airway hemangiomas (41).
Tracheal Obstruction
Tracheal Stenosis
Tracheal stenosis may be secondary to mucosal webs of variable thickness without any gross deformity of the underlying cartilage. Stenosis of the trachea can also be caused by complete tracheal rings of variable length. The segment with complete tracheal rings lacks the posterior membranous portion of the trachea.
Symptoms of airway obstruction from tracheal stenosis depends on the diameter of the narrowed airway lumen. Neonates with tracheal stenosis can present with persistent cough, respiratory distress, expiratory stridor, and wheezing. Symptoms tend to worsen following an upper respiratory infection, and sudden and complete obstruction can occur from mucus plugging of the narrowed segment. Many patients also tend to have feeding difficulties.
The diagnosis is suggested by history, airway films, and fluoroscopy; however, a definitive diagnosis is made by endoscopic evaluation. Mucosal web may respond to dilation alone. Short segments of tracheal stenosis are resected with end-to-end anastomosis (42). Long segments of stenosis require tracheoplasty using pericardial and/or cartilage grafts (43). These surgical procedures are associated with high morbidity and mortality rates.
The diagnosis is suggested by history, airway films, and fluoroscopy; however, a definitive diagnosis is made by endoscopic evaluation. Mucosal web may respond to dilation alone. Short segments of tracheal stenosis are resected with end-to-end anastomosis (42). Long segments of stenosis require tracheoplasty using pericardial and/or cartilage grafts (43). These surgical procedures are associated with high morbidity and mortality rates.
Tracheomalacia
In tracheomalacia there is increased flaccidity of the tracheal walls, which leads to anterior-posterior collapse of the airway and obstruction (44). Primary tracheomalacia is an isolated finding that usually resolves by age 2 years. Secondary tracheomalacia is seen in neonates with tracheoesophageal fistula (TEF) or external vascular compression. In primary tracheomalacia and in secondary tracheomalacia associated with TEF, deficiency in the tracheal cartilage may cause the abnormal collapse of the trachea (44,45).
Clinical symptoms of tracheomalacia range from mild to severe. Common features of severe cases include inspiratory and/or expiratory stridor, wheezing, persistent coughing, recurrent respiratory infections, reflex apnea, and difficulty with clearing airway secretions. Reflex apnea describes a respiratory arrest that can progress to a cardiac arrest and is seen in patients with tracheobronchial compression (46). Diagnosis of tracheomalacia can be suggested by fluoroscopy, but a definitive diagnosis can only be made by endoscopic examination of the airway under spontaneous ventilation. Widened semicircular cartilages, ballooning of the posterior membranous wall, and collapse of tracheal lumen are seen. In severe cases tracheotomy is required until the condition resolves. The use of positive airway pressure through the tracheotomy tube may be necessary to maintain patency of the airway below the tracheotomy tube (47).
Vascular Compression
Innominate artery compression of the trachea can occur when there is an anomalous distal origin of the innominate artery from the aortic arch. The innominate artery crosses the trachea in a left-inferior-to-right-superior direction and causes compression of the trachea. Infants with this condition present with stridor, recurrent respiratory infections, feeding difficulties, and reflex apnea. Diagnosis is best made by endoscopy and MRI. Surgery is necessary only in infants with severe stridor or reflex apnea. Suspension of the innominate artery through thoracotomy or median sternotomy and reimplantation of the innominate artery are available surgical options (48).
Other less common vascular anomalies causing airway obstruction include double aortic arch, right aortic arch with left ligamentum arteriosum, and pulmonary artery sling. Evaluation of these conditions includes chest radiographs, MRI, and endoscopy. In symptomatic infants, the appropriate surgical procedure should be undertaken by a cardiothoracic surgeon.
THORACIC LESIONS CAUSING RESPIRATORY DISTRESS
Congenital Lobar Emphysema
Congenital lobar emphysema can be severe or life-threatening as a result of hyperexpansion of a single lobe of the lung. Air is permitted into the involved lobe but denied egress. The lobe becomes emphysematous, resulting in compression of adjacent pulmonary parenchyma and mediastinal displacement. Symptoms may appear shortly after birth and invariably develop before 4 months of age (49). The cause is unknown. An inherent, cartilaginous defect in the bronchus has been postulated, but the bronchial abnormality is not always recognizable in the resected specimen.
A chest radiograph is characteristic, showing hyperaeration of the involved lobe with mediastinal shift away from the affected side. The lobar distribution of the hyperaeration can be appreciated, and adjacent pulmonary parenchyma is compressed. The upper lobes are most frequently involved, but the condition may be seen in the middle lobe. Rarely, the lesion is bilateral (50).
Treatment is surgical, and prompt thoracotomy and lobectomy are undertaken after the diagnosis is made. An infant may remain compensated for some weeks and then deteriorate rapidly from acute hyperinflation of the in-volved lobe. There is no place for expectant management of congenital lobar emphysema.
An identical clinical picture may be seen in the neonate who has been ventilated for a prolonged time and develops a large pneumatocele as a result of respiratory tract barotrauma. It is often difficult for the physician to determine whether respiratory distress is caused by the pneumatocele or by generalized pulmonary parenchymal disease. Surgical excision of the pneumatocele is indicated if the lesion is enlarging and the respiratory status is worsening without another apparent cause.
Cystic Adenomatoid Malformation
Cystic adenomatoid malformation (CAM) is a pulmonary maldevelopment that presents with cystic replacement of pulmonary parenchyma. If the cysts are small (i.e., microcystic CAM) and constitute a small portion of one lung, the child may be asymptomatic. If the lesion is microcystic and replaces a large portion of the lung, the fetus may develop hydrops, and the prognosis is poor (51). If the cysts are macroscopic and the child is in respiratory distress at birth, the problem may be misdiagnosed as congenital diaphragmatic hernia. Appropriate therapy for the symptomatic neonate with CAM is thoracotomy and resection of the involved lung. Postoperative support with extracorporeal membrane oxygenation (ECMO) has been necessary for
infants developing severe persistent pulmonary hypertension after CAM excision (52).
infants developing severe persistent pulmonary hypertension after CAM excision (52).
Bronchogenic Cyst
Bronchogenic cyst is another lung bud anomaly in which the normal bronchiole-to-bronchiole communication is absent or atretic, resulting in a mucus-producing cyst that may obstruct the trachea, bronchus, or esophagus. This seldom causes severe problems in the neonate, but it must be considered if a space-occupying lesion is detected on a chest x-ray film obtained for investigation of respiratory distress. Excision of the bronchogenic cyst is the preferred treatment.
Iatrogenic Airway Injury
As aggressive management of the pulmonary disease of newborns has developed, there has been a concurrent increase in injury to the airway or pulmonary parenchyma. Long-term intubation and high peak inspiratory pressure settings on the ventilator place these children at an increased risk of airway injury. It has been known since 1976 that perforation of bronchi, particularly the bronchus of the right lower lobe, can be prevented by careful measurement of suction catheters so that they do not extend more than 1 cm beyond the carina (53). The development of a bronchopulmonary fistula after chest tube evacuation of a pneumothorax can be life-threatening and may require surgical closure of the fistula, although some children have been successfully treated nonoperatively (54).
Complications of high-pressure ventilation include development of interstitial emphysema and bronchopulmonary dysplasia. The interstitial emphysema is usually self-limited, although some infants may benefit from a surgical approach to the problem (55). Surgery can be curative for granulomas within the airway of chronically intubated neonates, particularly if the lesions contribute to air trapping or stenosis. The best management for iatrogenic airway injury is prevention, and although not totally avoidable, careful attention to ventilator management and suction techniques should reduce the incidence and severity of these problems.
Diaphragmatic Hernia
Development of the diaphragm is generally complete the 12th week of gestation, by which time the bowel has returned to the abdominal cavity. Failure of the development of the posterolateral portion of the diaphragm results in persistence of the pleuroperitoneal canal or foramen of Bochdalek, which allows the viscera to occupy the chest cavity, and the abdomen is underdeveloped and scaphoid. Both lungs are hypoplastic, more so on the side of the hernia. Bronchial branching, lung weight, and lung volume are decreased. The pulmonary arteries are hypoplastic (56,57). The lesion occurs in one of every 3,000 live births, with equal frequency in male and female infants. Sporadic occurrence is the rule, but a familial pattern has been reported (58,59).
The prenatal diagnosis of congenital diaphragmatic hernia (CDH) can be made using fetal sonography as early as 15 weeks of gestation (60). Sonographic findings include herniated abdominal viscera, abnormal anatomy of the upper abdomen, and mediastinal shift away from the herniated viscera (61). The high-risk fetus is identified by a diagnosis early in gestation, a dilated stomach in the chest, low lung-to-thorax ratio, low lung-to-head ratio, and polyhydramnios. Amniocentesis with a fetal karyotype identifies chromosomal defects; trisomy 18 and 21 are most common. More than 40% of newborns with CDH have associated anomalies of the heart, brain, limbs, genitourinary system, or craniofacial region (62).
Typically, babies with congenital Bochdalek hernia present dramatically with cyanosis (See Color Plate) and severe respiratory distress immediately after birth. Because the abdominal viscera are dislocated through the defect into the chest, the abdominal contour is scaphoid. Breath sounds are diminished or absent, and because the mediastinal structures have been displaced, the heart sounds are heard in the right chest. As the bowel fills with gas, respiration and cardiac action are further compromised, hypoxia and respiratory acidosis are increased, and death is inevitable unless appropriate intervention is undertaken. Congenital diaphragmatic hernia is no longer considered a surgical emergency unless the viability of the herniated bowel is in question. In some instances, the infants remain relatively asymptomatic in the early hours and days of life, and rarely, a diaphragmatic hernia is an incidental finding in an older child. The respiratory symptoms demand an immediate x-ray film, which is diagnostic. The hernia is on the left side in 90% of these infants, and the air-filled bowel is seen occupying the left hemithorax, with resultant displacement of the mediastinum to the right (Fig. 44-4). The abdomen is gasless. Additional x-ray films are unnecessary, and contrast studies for additional confirmation are contraindicated.
Many newborns with CDH have respiratory failure within minutes of birth, and urgent stabilization is mandatory to reverse hypoxia, hypercarbia, and metabolic acidosis. Prompt and aggressive preoperative care is essential (63,64). This generally includes mechanical ventilation with 100% oxygen, sedation with narcotics, muscle paralysis, controlled alkalosis with hyperventilation and intravenous sodium bicarbonate, and vasopressors. Permissive hypercarbia and gentle ventilation have proven effective in a number of centers. Regardless of the mode of therapy, the goal is to reverse the baby’s persistent pulmonary hypertension with right-to-left shunting of oxygen-poor blood across the open foramen ovale and the ductus arteriosus.
Some infants do not improve despite aggressive therapy, and many centers use ECMO before hernia repair to stabilize these desperately ill infants (65,66). Venovenous or venoarterial bypass is used, depending on the infant’s hemodynamic stability. Bypass is continued until the pulmonary hypertension is reversed and lung function is
improved. Most infants respond within 7 to 10 days, but some require up to 3 weeks of support. Newborns who have not improved after this time probably have such severe pulmonary hypoplasia that further extracorporeal life support is futile.
improved. Most infants respond within 7 to 10 days, but some require up to 3 weeks of support. Newborns who have not improved after this time probably have such severe pulmonary hypoplasia that further extracorporeal life support is futile.
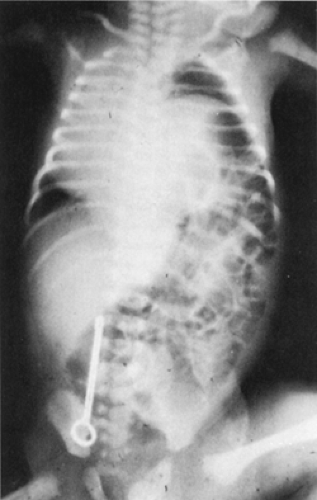 Figure 44-4 X-ray film of left diaphragmatic hernia with loops of bowel well up into the chest. Although most diaphragmatic hernias do not have a sac, the smooth curve of the sac in this instance is visible. Notice that the heart is displaced to the border of the right chest. |
There are no absolute respiratory criteria that exclude newborns with CDH from consideration for ECMO support (67,68). Approximately 60% of infants with CDH who are supported by ECMO are expected to survive (see Chap. 32).
The surgical findings are usually those of a posterolateral defect in the left diaphragm, with most or all of the abdominal viscera in the chest. The surgeon reduces the hernia gently by withdrawing the viscera from the chest. If a sac is present, it is delivered and excised. There may be adequate diaphragmatic tissue to accomplish direct suture repair. If a significant portion of the diaphragm is lacking, prosthetic material is used to close the defect. Before completion of the repair, a small chest tube may be placed in the left hemithorax and brought out through an intercostal space.
The temptation to expand the compressed lung at the time of initial surgery must be resisted. Aggressive attempts at expansion can result in pneumothorax on the contralateral side, which, if unrecognized, is a disastrous complication.
The abdominal viscera have been located in the thorax throughout most of the developmental period of the fetus; thus, there is insufficient room within the abdomen to accommodate the intestine without dangerously increasing the intraabdominal pressure, compressing the vena cava, and compromising respirations by elevating the diaphragm. To avoid these potential problems, the surgeon may omit anatomic closure of the abdominal wall. Skin flaps are quickly mobilized, and only the skin is closed, or the abdomen is closed by creating a silastic silo as for gastroschisis or a large omphalocele. The ventral pouch created accommodates the intraabdominal organs; diaphragmatic action and venous return are unimpeded. The ventral hernia is repaired after the infant has been weaned off the ventilator and is in stable clinical condition.
Ten percent of infants with Bochdalek hernias present with the defect on the right side. The difference in incidence on the two sides may be explained by the presence of the liver, which partially blocks the pleuroperitoneal canal and limits the amount of bowel that can herniate into the chest. Symptoms in babies with right-sided hernias may be less severe, but when a right diaphragmatic hernia of Bochdalek presents as an emergency, it is managed as described.
After CDH repair and removal from ECMO, ventilation is continued with ventilation rates and oxygen concentrations necessary to maintain adequate oxygenation. Continuous transcutaneous oxygen monitoring of upper and lower body areas is useful. To avoid relapse into pulmonary hypertension, weaning from the ventilator should be achieved by making small incremental changes in the inspired oxygen and ventilator rate.
New modalities being investigated may offer an increased chance of survival for infants with CDH. These include surfactant replacement therapy, liquid ventilation, intratracheal pulmonary ventilation, and pulmonary lobar transplantation (69,70). Prenatal repair of the diaphragmatic hernia has been abandoned, as there was no improvement in survival or morbidity in a randomized trial. Current prenatal therapy of CDH is directed at occluding the trachea, which results in enlargement of the lungs with retained fluid. The baby is then delivered by planned cesarean section, at which time the trachea is repaired or intubated, with the baby remaining on placental support. Initial discouraging results with this technique have been improved with recent modifications; however, fetal tracheal plugging remains an experimental procedure that is being evaluated in a limited number of centers (71,72).
Babies who present after the first day of life with signs and symptoms prompting a diagnosis of diaphragmatic hernia are almost always hardier patients and have greater pulmonary reserve, and they can be expected to make a satisfactory recovery.
The anterior retrosternal hernia of Morgagni is rarely encountered in the newborn. The diagnosis is confirmed by chest radiograph in the lateral projection. The standard treatment is surgical reconstruction, beginning with an abdominal approach through a thoracoabdominal
incision. The prognosis is usually favorable, and these lesions are not associated with the severe cardiopulmonary complications seen with Bochdalek hernias in the neonatal period.
incision. The prognosis is usually favorable, and these lesions are not associated with the severe cardiopulmonary complications seen with Bochdalek hernias in the neonatal period.
Eventration of the Diaphragm
Eventration of the diaphragm may be congenital or acquired. The congenital presentation may mimic that of a congenital diaphragmatic hernia with a sac. The acquired lesion results from paralysis of the diaphragm, most commonly caused by operative trauma or birth injury (73).
Large eventrations and diaphragmatic paralysis are poorly tolerated by infants (74). Paradoxic cephalad motion of the diaphragm on inspiration produces a shift of the mobile, neonatal mediastinum that limits the function of the contralateral lung. Moderate or severe respiratory distress is evident; many newborns with eventration require ventilatory support.
Diagnosis is suggested by a marked elevation of a hemidiaphragm on a chest radiograph. Fluoroscopic examination identifies paradoxic movement of the diaphragm. Treatment is plication of the diaphragm with nonabsorbable sutures that reef up or overlap the diaphragm. The taut diaphragm results in less abnormal motion and improved ventilation.
LESIONS OF THE ESOPHAGUS
Esophageal Atresia and Associated Anomalies
The success story of the management of infants born with esophageal atresia and tracheoesophageal fistula is one of the most dramatic and satisfying that the pediatric surgeon, the neonatologist, and the pediatrician can point to. In the early 1900s, virtually all babies born with esoph-ageal atresia and tracheoesophageal fistula died. In 1941, Haight and Towsley were the first to bring an infant with esophageal atresia and tracheoesophageal fistula successfully through the rigors of primary transthoracic reconstruction (75). This landmark accomplishment occurred before antibiotics, respiratory support, or sophisticated intravenous nutrition were available. This surgical ap-proach formed the basis of modern operative and postoperative care of infants born with this anomaly. Fifty years after the first survivor was announced, every baby born with atresia of the esophagus who is spared coexisting fatal abnormalities and is offered appropriate care has an excellent chance of leading a normal life.
Embryologic and Genetic Considerations
The cause of esophageal atresia is unknown, but it is related to the common origin of the esophagus and trachea (76). The embryonic trachea and esophagus are first recognized as a ventral diverticulum of the foregut approximately 22 or 23 days after fertilization (77). As the diverticulum elongates, a proliferation of endodermal cells appears on the lateral walls. These cell masses become ridges of tissue that ultimately divide the foregut into tracheal and esophageal channels. The division into separate tubes is completed between 34 and 36 days after fertilization. Many embryologic studies indicate that interruption of development in the fourth fetal week allows persistence of fistulas and clefts between the esophagus and trachea and permits incomplete development of the esophagus.
There are reports of familial occurrences of esophageal atresia, which raises the possibility of a heritable genetic factor. Numerous accounts of twins and siblings with esophageal atresia have been reported (78). Conversely, certain commonly coexistent anomalies, such as the VACTERL (i.e., vertebral, anal, cardiac, tracheal, esophageal, renal, and limb anomalies) association (see Chapter 38) and other malformations, strongly suggest that the developing fetus is affected by a teratogen or defect of embryogenesis (79).
In many babies with esophageal atresia, it is the associated anomalies that alter treatment and affect survival. Congenital heart defects and chromosomal abnormalities are the most worrisome. Major anomalies that may seriously affect the infant but that are not usually fatal include imperforate anus and other congenital obstructions of the gut. Grosfeld and Ballantine found that 31 (37%) of 84 infants had cardiac anomalies; 18 (21.4%) had GI malformations, of whom 11 (13%) had imperforate anus; and six (7%) had the VACTERL association (80). A ventricular septal defect is the most common cardiac lesion, followed in frequency by patent ductus arteriosus and tetralogy of Fallot. Piekarski and Stephens suggest that the high incidence of coexisting anomalies is a reflection of generalized damage to the mesenchymal tissue in the fourth week of gestation (81).
Esophageal Atresia with Tracheoesophageal Fistula
Esophageal atresia occurs in approximately one of 3,000 to 4,500 births. In the most common form of esophageal anomaly (86% of patients), the blind-ending upper esophageal segment usually extends into the upper portion of the thorax, and the lower portion of the esophagus is connected to the trachea at or just above the tracheal carina (Fig. 44-5) (82). This connection is 3 to 5 mm in diameter and easily admits inspired air or, in a retrograde fashion, acidic gastric secretions. The earliest clinical sign of esophageal atresia is excessive oral secretions or regurgitation of saliva. The saliva pools in the blind-ending esophagus and then accumulates until it is apparent around the lips as excessive mucus. The first feeding is followed by choking, coughing, and regurgitation. Abdominal distension is a prominent feature, occurring as inspired air is transmitted through the fistula and distal esophagus into the stomach. Gastric juice may pass upward in the distal esophagus, traversing the tracheoesophageal fistula and spilling into the trachea and lungs, leading to chemical
pneumonia. Pulmonary difficulties are compounded by atelectasis and diaphragmatic elevation secondary to gastric distension.
pneumonia. Pulmonary difficulties are compounded by atelectasis and diaphragmatic elevation secondary to gastric distension.
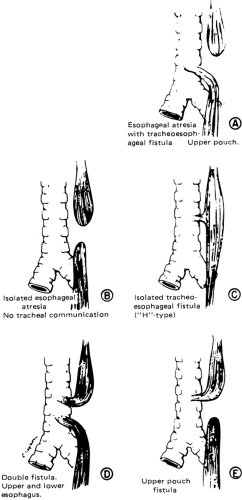 Figure 44-5 The various forms of esophageal malformations are shown in the order of the frequency in which they occur. From Nardi GL, Zuidema GD. Surgery, 3rd ed. Boston: Little, Brown, 1972, with permission. |
Diagnosis
The diagnosis of many infants with esophageal atresia is made prenatally. Polyhydramnios is the most frequent finding, particularly in infants with pure esophageal atresia. Those with a tracheoesophageal fistula may not develop polyhydramnios if the communication is large enough to permit swallowing of the amniotic fluid through the fistula. An additional finding on a prenatal ultrasound is failure to identify the fetal stomach, which should raise the suspicion for esophageal atresia. The prenatal diagnosis of esophageal atresia permits the search for other anatomic anomalies and possible chromosomal defects. Appropriate prenatal counseling and preparation of the family can be initiated, specifically with arrangement for postnatal correction, unless contraindicated by coexistent abnormalities.
Once the infant is born, attentive members of the nursing staff who are feeding the baby are often the first to suspect the esophageal blockage. Esophageal atresia may not be obvious on the initial newborn examination unless an attempt is made to pass a tube into the stomach. Thin, flexible feeding catheters should be avoided because they may coil up in the esophagus and give the misleading impression that they have passed into the stomach. A larger, stiffer catheter carefully advanced will meet the obstruction. Occasionally, a tube dissects into the wall of a normal esophagus, leading to a misdiagnosis of esophageal atresia, particularly in a premature infant. A contrast x-ray film rules this out and confirms the diagnosis of atresia; a lateral projection with 1 mL of dilute barium or an isoosmolar contrast agent (e.g., metrizamide) shows the length of the upper pouch, defines its precise extension into the chest, and demonstrates the rare upper pouch fistula (Fig. 44-6A,B). Air seen in the bowel confirms the presence of a tracheo-esophageal fistula. The existence of pneumonia or atelectasis also can be demonstrated on the initial radiographs.
Evaluation of the heart and great vessels with echocardi-ography is important to identify potential cardiac anomalies and verify the aortic arch position. A right-sided arch may alter the surgical approach and exposure. Bronchoscopy is useful to identify the level of the fistula and to exclude upper pouch fistulas and laryngotracheoesophageal clefts.
Management
After the diagnosis is secure, the following measures should be instituted promptly:
Basic supportive measures, such as an infant warmer.
Place the infant in a 30- to 40-degree head-up position.
Give nothing orally.
Provide intravenous antibiotics for possible aspiration.
Place a sump suction catheter in the upper pouch to remove the excess secretions (Fig. 44-7).
Consult with the appropriate pediatric surgical service.
The traditional approach to the timing of surgical repair was based on the increased risk of operating on infants with low birth weight and pneumonia. However neonatal care has evolved to the point that neither low birth weight nor the presence of pneumonia is a risk factor for poor survival (83). Severe anomalies and their consequences are now the crucial determinants of survival. Therefore, at Children’s National Medical Center (CNMC), each baby admitted with esophageal atresia is managed according to
his or her physiologic status alone (84). If the infant is stable, immediate primary repair is undertaken. If unstable, surgery is delayed until the clinical status is stabilized, the impact of associated anomalies is determined, and the infant can be anesthetized and operated on safely.
his or her physiologic status alone (84). If the infant is stable, immediate primary repair is undertaken. If unstable, surgery is delayed until the clinical status is stabilized, the impact of associated anomalies is determined, and the infant can be anesthetized and operated on safely.
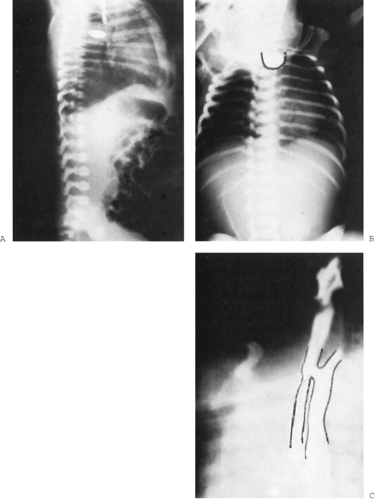 Figure 44-6 A: Lateral radiograph of a baby with esophageal atresia and tracheoesophageal fistula reveals a small meniscus of barium in the upper pouch. Gas is present in the stomach and intestinal tract because of the fistulous connection to the trachea. In this radiograph, some air in the lower esophageal segment can be seen in the posterior mediastinum. B: In a radiograph of a patient with isolated esophageal atresia, the upper pouch is outlined by barium. There is no air below the diaphragm. C: In a barium swallow in a patient with H-type isolated tracheoesophageal fistula, a normal-sized lumen of the esophagus is seen. Dye has spilled into the trachea, outlining the upper trachea and larynx. The fistula is at the level of the clavicle. |
Of historic interest, a classification developed by Waterston and colleagues in 1962 was useful in the stratification of patients for different management plans and the comparison of outcomes (85). The infants were classified as follows:
Category A: Birth weight over 2.5 kg (5.5 lb) and otherwise well
Category B: Birth weight of 1.8 to 2.5 kg and well, or higher birth weight but moderate pneumonia and other congenital anomalies
Category C: Birth weight under 1.8 kg, or higher birth weight but severe pneumonia and severe congenital anomaly.
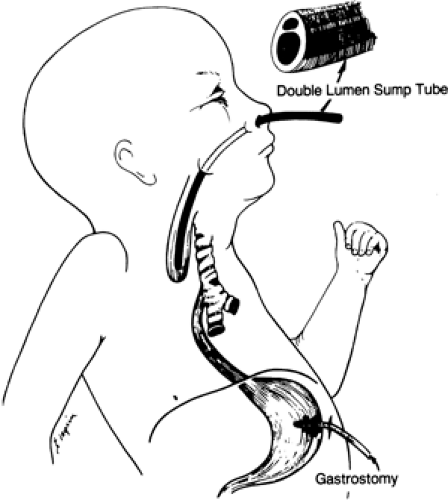 Figure 44-7 Temporary care of a patient with esophageal atresia and tracheoesophageal fistula is performed with the patient in the upright position. A gastrostomy is in the stomach, and a double-lumen sump tube is in the upper pouch to clear secretions and saliva. |
Immediate Operative Repair
A thoracic incision provides exposure of the upper pouch and tracheoesophageal fistula. A right thoracotomy is standard unless the aortic arch is on the right, which would interfere with the dissection. A retropleural approach affords protection of the lung by maintaining its pleural envelope. If an anastomotic leak occurs, it will not communicate with the pleural cavity but can be drained posteriorly from the mediastinum with less morbidity.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


