17 Surgery
Head and Neck
Introduction
Most lesions of the head and neck are benign in nature. Lesional location provides essential information about the probable diagnosis. Table 17-1 provides a summary of the common pediatric head and neck lesions by anatomic location. Physical examination and diagnostic imaging studies are important to generate a differential diagnosis and further determine the nature and extent of the lesion. Critical physical examination findings include determination of size, evidence of airway compromise, signs of inflammation, presence of sinus tracts, or ocular involvement.
Table 17-1 Common Lesions of the Head and Neck in Infancy and Childhood
| Region | Location | Common Lesions |
|---|---|---|
| Head | Scalp | Hemangioma, dermoid cyst |
| Ear | Preauricular sinus, tag | |
| Eyebrow | Dermoid cyst | |
| Base of nose | Meningocele, encephalocele | |
| Parotid gland | Hemangioma, lymphangioma, rhabdomyosarcoma, lymphoma, mixed tumor, parotitis | |
| Mouth | Tongue | Tongue-tie, macroglossia, lingual thyroid |
| Floor of mouth | Ranula | |
| Cheek and lip | Papilloma, mucocele | |
| Alveolar ridge | Tooth bud, epignathus | |
| Neck | Midline | Thyroglossal duct cyst, dermoid cyst, submental lymph node, goiter |
| Lateral | Branchial cleft cyst or sinus, lymphadenitis, lymphoma, lymphangioma, torticollis |
Cervical Lymphadenopathy
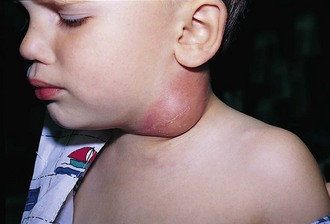
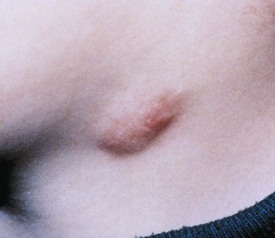
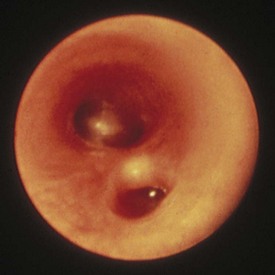
Benign reactive cervical lymphadenopathy is the most common mass in the lateral triangle of the neck. These lesions arise as nonspecific hyperplastic responses to infection of the upper respiratory tract (nose, sinuses, ears, mouth, and pharynx) or skin (face and scalp). Typically these nodes are less than 2 cm in size and are rubbery, oval, and isolated. They characteristically occur in children between 2 and 10 years of age. Streptococcus pyogenes and Staphylococcus aureus are the most common organisms that produce this adenopathy. In most instances, the nodes spontaneously regress after resolution of the inciting infection. Bacterial infection within node(s) may lead to more significant enlargement with increased tenderness, erythema, and ultimately suppuration (Fig. 17-1). Aggressive antibiotic therapy in the early stages of infection may prevent the development of the late suppurative stages that require surgical intervention. Fluctuant masses should be aspirated or incised and drained.
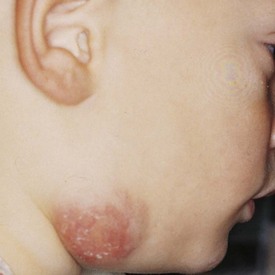
Various clinical presentations may occur with mycobacterial infections including local cervical adenopathy, pulmonary infection, and disseminated disease. The most common form is caused by one of the Mycobacterium avium-intracellulare-scrofulaceum (or MAIS) complex, which consists of approximately 15 organisms. These mycobacterial organisms typically produce local cervical disease. Mycobacterial tuberculosis usually presents with pulmonary infection but may rarely have supraclavicular or cervical lymphadenopathy (Fig. 17-2). In contrast, atypical mycobacterial infection usually involves the submandibular, submaxillary, or preauricular lymph nodal regions. Large, firm, immobile, and nontender lymph nodes may arise after inoculation. These may undergo spontaneous breakdown with the development of an abscess and sinus formation. Incision and drainage of fluctuant nodes may lead to a chronically draining sinus.
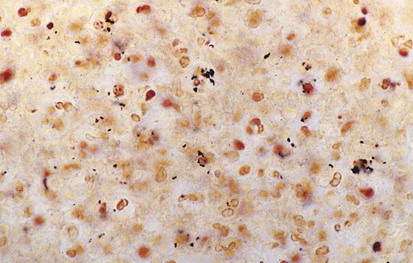
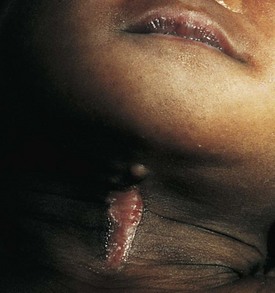
The differential diagnosis of chronic cervical lymphadenopathy (i.e., nodes that persist beyond 4 weeks) includes cat-scratch disease, atypical mycobacterial infection, and tuberculosis. Cat-scratch disease, a common cause of lymphadenopathy in children, usually develops as a regional nodal enlargement 2 to 4 weeks after inoculation by either a dog or cat. There may be a local reaction to the scratch followed by the evolution of lymphadenopathy, which may persist for several months. On occasion these nodes become suppurative and fluctuant and require surgical drainage. The diagnosis may be made by serologic testing for the antigen or by polymerase chain reaction of nodal tissue. Alternatively, Warthin-Starry, a histochemical silver stain for cat scratch, may identify the Bartonella organisms in tissue specimens (Fig. 17-3). Complete resection of the node and any tracts is usually curative. Antimycobacterial therapy is not indicated in most cases of MAIS infection.
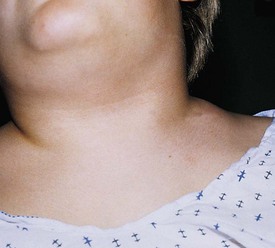
Cervical adenitis secondary to Mycobacterium tuberculosis infection is usually a manifestation of significant intrathoracic disease and requires aggressive antimycobacterial drug therapy. Surgery is usually unnecessary and should be avoided because of the risk of developing a chronically draining sinus (cervical tuberculosis). Lymphoma may present as painless cervical adenopathy. The absence of antecedent upper respiratory or cutaneous infections, the persistence of nodes beyond 6 weeks, size greater than 2 cm, and firm consistency should raise concern for malignancy. Although cervical adenopathy is more common in Hodgkin disease, non-Hodgkin lymphoma may also present with a cervical mass (Fig. 17-4). Incisional biopsy is diagnostic and mandated when these criteria are met. Other primary malignancies such as neuroblastoma and rhabdomyosarcoma may present as lateral neck masses. Secondary metastases from intraabdominal or head and neck tumors may also occur.
Thyroglossal Duct Cyst and Sinuses
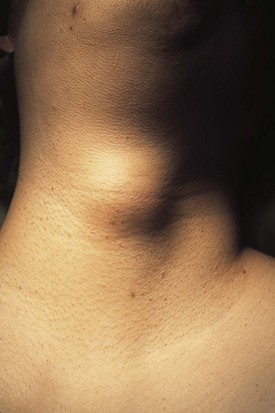
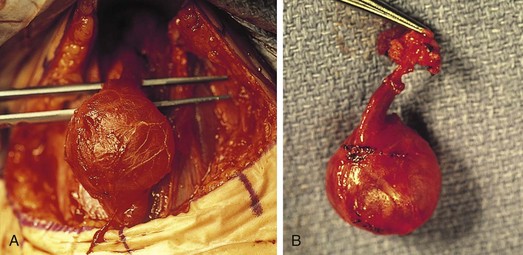
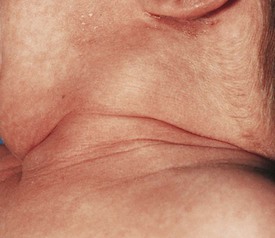
The differential diagnosis for midline neck masses includes thyroglossal duct remnants, dermoid cysts, and lymphadenopathy. During fetal development, the thyroid gland originates at the base of the foramen cecum and descends in the midline along the course of the thyroglossal duct close to the hyoid bone, until it reaches its final destination at the base of the neck. Failure of regression of the thyroglossal duct may lead to cyst formation (Fig. 17-5). These lesions are quite prone to infectious complications and require surgical excision. This excision requires resection of the midportion of the hyoid bone and ligation of the tract leading to the foramen cecum to prevent future recurrence (Fig. 17-6).
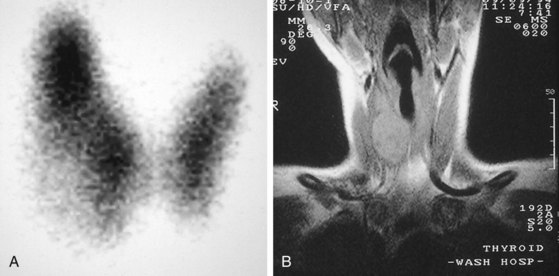
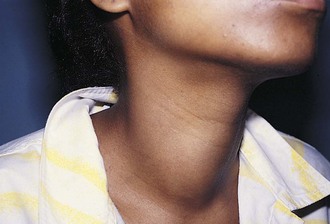
Thyroid nodules are common in the pediatric population (Fig. 17-7). However, a greater incidence of malignancy occurs within these lesions in children. Therefore, thorough evaluation and management of these lesions are critical to a favorable outcome. These nodules are twice as common in girls as boys. They typically present with a midline anterior cervical mass that moves with the thyroid gland. Initial physical examination determines the location of the lesion, as well as the presence of associated lymphadenopathy. In general, clinical findings are unreliable in distinguishing between benign and malignant disease. Although thyroid imaging studies are often indicated, they are rarely helpful in aiding with the diagnosis, unless there is evidence of multiple nodules. A multinodular goiter would suggest nodular Hashimoto disease, which is the most common benign lesion of the thyroid (Fig. 17-8). The utility of ultrasound in distinguishing benign from malignant disease on the basis of a cystic appearance of the lesions is also unhelpful. Furthermore, the utility of fine-needle aspiration cytology in the pediatric population remains an area of considerable debate; reports in the older medical literature suggested a much higher incidence of malignancy in thyroid nodules in children than do current studies. Consequently, there was a much stronger recommendation for surgical excision as the diagnostic procedure of choice. More recent reports have shown that the incidence of benign disease is approximately 20%. Therefore needle aspiration may avert surgical resections for benign disease. Needle aspiration cytology that shows a true malignancy or that has indeterminate pathology should be followed by surgical resection. Total thyroidectomy is recommended for malignant primary lesions; lobectomy and isthmusectomy are recommended for benign lesions in which cancer cannot be completely ruled out.
Other miscellaneous conditions may occur in the anterior neck, such as midline branchial (cervical) cleft, a linear tract of epithelialized tissue in the anterior midline of the neck that occurs because of aberrant fusion of the branchial arches (Fig. 17-9). In addition, mediastinal lesions such as thymic cyst may present as midline cervical masses (Fig. 17-10).
Branchial Cleft and Arch Anomalies
Cysts and Sinuses
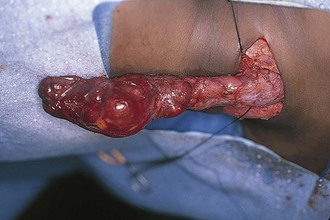
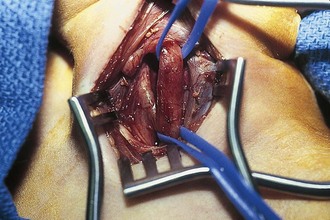
Branchial cleft anomalies give rise to cysts and sinuses in the lateral triangle of the neck. Second branchial cleft anomalies are the most common and typically present as an opening along the lower anterior border of the sternocleidomastoid muscle. These sinuses have their origin in the tonsillar fossa and may travel between the carotid sheath to exit along the border of the sternocleidomastoid muscle. A complete fistula drains through a sinus opening (Fig. 17-11), whereas an incomplete fistula may present as a simple cystic structure in the subcutaneous tissue in the region (Fig. 17-12). Secondary infection of these lesions is common. Excision of the tract to the site of origin in the peritonsillar region prevents recurrence. Other branchial cleft and arch anomalies are less common (Fig. 17-13).
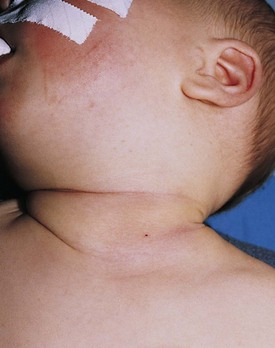
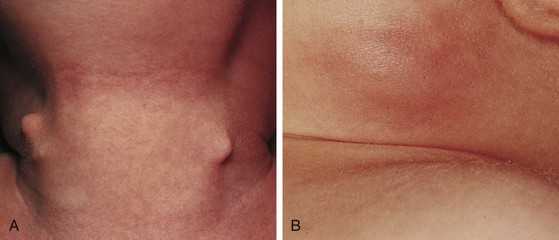
Fibromatosis coli, or fibrous dysplasia of the sternocleidomastoid muscle, is commonly seen in infants and young children (Fig. 17-14). These children present with a mass in the lower neck with tilting of the head and face to the side of the lesion (Fig. 17-15). Parents most often bring their infants in because they are concerned about the possibility of a malignant tumor, whereas older children may present with hemifacial hypoplasia and asymmetry. Early recognition of this condition in infancy and the institution of daily physical therapy may avert surgery and long-term cosmetic deformity. Plagiocephaly and facial asymmetry are the sequelae of untreated deformities.
Scalp and Face Lesions
Scalp
Hemangiomas are benign, congenital, vascular tumors that most frequently arise in the head and neck. These lesions are typically raised above the skin level and may be red or somewhat purple in color. They may blanch on contact. Often hemangiomas may not be present at birth but develop in the first few months of life. Rapid growth and expansion may occur, leading to platelet sequestration and coagulopathy, known as the Kasabach-Merritt syndrome. This condition may be refractory to various medical maneuvers including steroids, radiation therapy, and chemotherapy. Despite significant growth in size during the first year of life, most hemangiomas have a benign course and undergo spontaneous resolution over the first 7 years of life (Fig. 17-16; and see Chapter 8). Surgical intervention is rarely necessary and should be reserved for those patients with impending airway compromise or periorbital involvement. Special consideration must be given to those patients with lesions extending toward the eye or impinging on the airway. Subsequent blindness or airway compromise may be the consequences if intervention is delayed.
Figure 17-16 Involuting parotid and neck hemangioma. Note the grayish discoloration indicative of resolution.
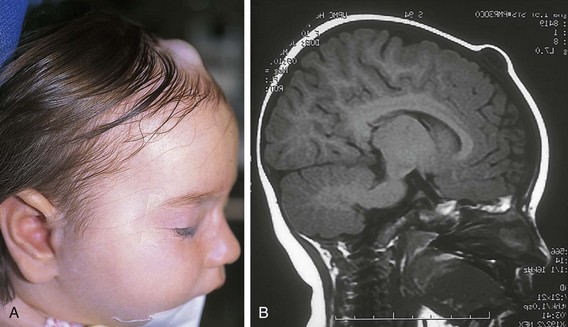
Dermoid cysts are congenital lesions that are composed of sequestered hair, skin, and sebaceous structures that occur in areas of embryonic fusion. These lesions are most frequent in the head and neck, but they may arise in other midline sites including the sacral, perineal, and sternal region. They are typically located in the head and neck along the lateral palpebral fissure, occipital scalp, and midline of the neck. Scalp dermoids are often well circumscribed, firm, and fixed to underlying deep structures such as bone, typically arising from the outer bony table of the skull. Midline scalp or back dermoids may have intracranial or intraspinal extension, respectively. They should always be evaluated by MRI before surgical intervention. Dural or central nervous system extension mandates neurosurgical consultation before resection. Treatment for these conditions is surgical (Fig. 17-17).
Face
Surgically significant salivary gland pathology is uncommon in the pediatric population. Hemangiomas are the most common benign lesions of the parotid gland in children. As with hemangiomas in other sites, these may not be entirely visualized at birth and may occur over the first few months of life. A small cutaneous birthmark may be the only initial presentation. Rapid growth and significant asymmetry may become apparent (Fig. 17-18). Fortunately, these lesions spontaneously involute with time. In the absence of early complications, surgery, sclerotherapy, or intralesional injection techniques should be reserved until the period after involution. Other causes of parotid enlargement in children include viral (mumps), bacterial (staphylococcal), and mycobacterial (atypical mycobacterial infection or tuberculosis) infections, as well as chronic inflammatory conditions. Treatment specific to these conditions is indicated.
Various intraoral lesions may arise that have surgical significance primarily because of their effects on swallowing, speech, and breathing. Tongue-tie (ankyloglossia inferior) occurs commonly in infancy, usually resolving spontaneously. Some cases persist, and these children may have impaired speech development. Usually a thin membranous structure, the frenulum, regresses with feeding. A persistent frenulum may impair speech and feeding. Simple division is therapeutic. Similarly, ranulas may form as pseudocysts in the floor of the mouth. Some spontaneously resolve, whereas a few may become quite large and impair lingual mobility, feeding, speech, and most significantly, breathing. Marsupialization or complete excision is curative (Fig. 17-19).
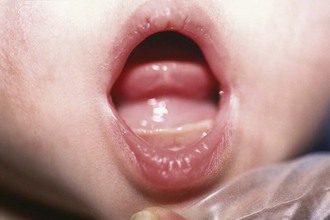
Figure 17-19 A ranula arises in the floor of the mouth, caused by congenital obstruction of the sublingual duct.
Lymphangiomas of the floor of the mouth may pose especially challenging management problems (Fig. 17-20). These lesions may cause significant macroglossia that obstructs the airway, requiring tracheostomy. Small vesicular lesions may occur on the lingual surface and exude fluid that may become purulent. Suppurative glossitis may require systemic antibiotic therapy. In addition to airway complications, problems with speech development and mandibular growth may occur. Some authors have proposed partial glossectomy as a therapy. More recent therapies for large intraoral and cervical lymphangiomas extending to the floor of the mouth involve sclerosant injection (hypertonic saline, alcohol, or OK-432) to avoid the morbidity and disfigurement associated with surgery in these areas.
Chest
Introduction
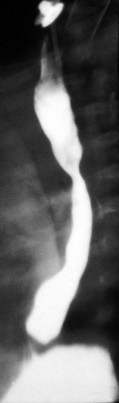
The clinical evaluation of these patients should include both anteroposterior (AP) and lateral radiographs of the chest. Particular focus should be directed to the soft tissue views of the neck, mediastinum, and airway contour. Fluoroscopic examination provides critical insight into the airway contour and diaphragmatic mobility throughout the respiratory cycle. Esophagogram with either barium or water-soluble contrast may be useful to delineate a vascular ring or mediastinal mass (Fig. 17-21). More invasive studies such as upper airway and esophageal endoscopy may be necessary to elucidate other surgical causes of respiratory distress. Surgical causes of respiratory distress may be subclassified into three major categories: upper airway, intrathoracic, and extrathoracic.
Upper Airway
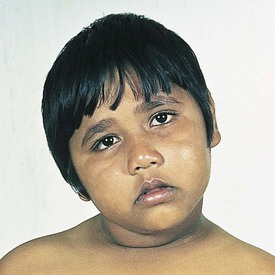
The initial evaluation of infants with presumed airway obstruction should include the passage of a nasogastric tube. Signs of pharyngeal obstruction suggest choanal atresia. This obstruction may be membranous (90%) or bony (10%). Half of these patients may have other forms of associated craniofacial or remote congenital anomalies that require concurrent evaluation and management. Nasopharyngoscopy is diagnostic in most cases. The oral airway must be maintained, and the baby must be fed via gavage feedings until transpalatal repair. Oropharyngeal obstruction may be caused by macroglossia or jaw bony abnormalities. Beckwith-Wiedemann syndrome is associated with lingual hypertrophy and gigantism (Fig. 17-22). Presentation in the newborn should alert the practitioner to the possibility of hypoglycemia secondary to hyperinsulinism. Permanent neurologic sequelae may result from diagnostic delay. Sublingual or lingual lymphangiomas may be associated with massive macroglossia that leads to airway distress. The hypoplastic and recessed mandible associated with Pierre Robin syndrome may cause a normal-sized tongue to fall posteriorly and obstruct the airway (Fig. 17-23). The association of cleft palate and cardiac defects with Pierre Robin syndrome may further exacerbate respiratory distress. Prone positioning of the infant may assist breathing and avert the need for tracheostomy. Alternatively, tracheostomy placement may provide a safer temporizing measure to allow adequate mandibular growth and development and to prevent obstruction. Newer techniques of mandibular distraction may avoid the need for prophylactic tracheostomy.
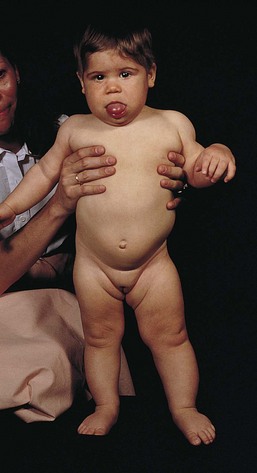
Figure 17-22 Beckwith-Wiedemann syndrome. Note hemihypertrophy on the left side, along with the prominence of the tongue.
(Courtesy D. Becker, MD, Pittsburgh, Pa.)
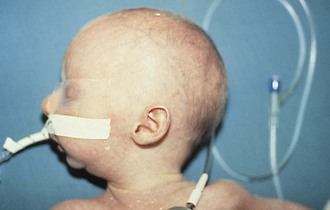
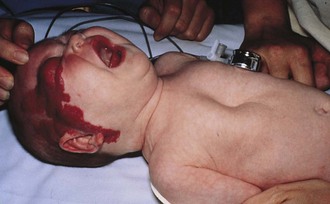
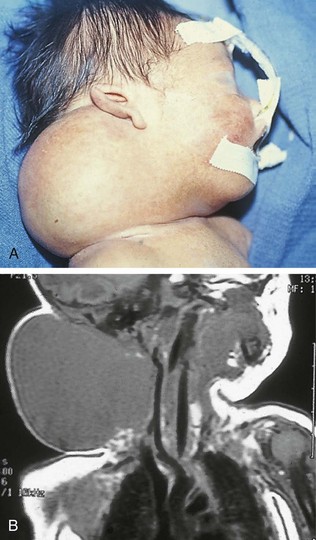
Pharyngeal masses may be another source of upper airway obstruction. These masses include branchial cleft remnants, dermoids, pharyngeal duplications, hemangiomas, lymphangiomas, lingual thyroids, sublingual teratomas, and Zenker diverticula. Large cervical masses such as a cystic hygroma (lymphangioma) (Fig. 17-24) and cervical teratoma (Fig. 17-25) may induce airway compression and cause dyspnea. Antenatal diagnosis of the lesions may indicate a potential airway emergency. The ex utero intrapartum treatment (EXIT) procedure allows time to secure airway control before division of the umbilical cord (Fig. 17-26).
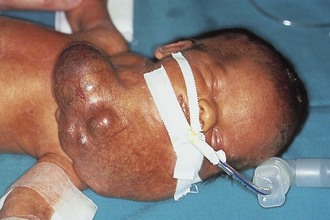
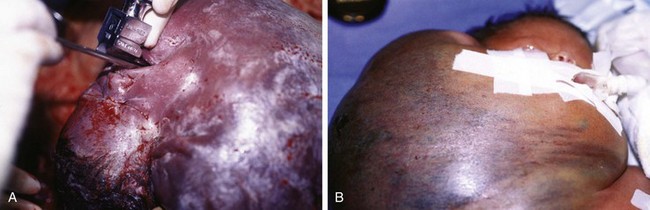
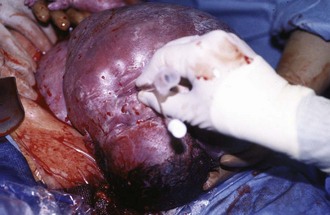
Figure 17-25 A, Cervical teratoma at delivery presents airway challenges (B) in the perinatal period.
Mediastinum and Diaphragm
Mediastinal masses are uncommon in the pediatric population. The limited space of the thoracic cavity predisposes normal structures to be compressed by space-occupying lesions. Masses in this anatomic region may lead to numerous symptoms including cough, dysphagia, dyspnea, and rarely, superior vena cava syndrome. The mediastinal location and the patient’s age provide the most critical insight into the diagnosis. The mediastinum may be divided into three major compartments: anterior–superior, middle, and posterior. The location of a mass in any one of these compartments is an important diagnostic feature (Table 17-2). The anatomic boundaries for these compartments include the sternum to the anterior aspect of the trachea and pericardium (anterior); the trachea, major bronchi, and paratracheal structures (middle); and the posterior aspect of the trachea to the spine (posterior). Anterior mediastinal masses typically arise in tissues of thyroid or thymus, and are lymphoid or teratomatous in origin (Fig. 17-27). Middle mediastinal masses are typically tumors or congenital anomalies arising from the tracheobronchial tree, lymph nodes, esophagus, or pulmonary parenchyma. Posterior mediastinal masses are primarily neurogenic tumors or congenital enterogenous lesions (Fig. 17-28).
Table 17-2 Mediastinal Masses in Childhood
| Anterior–Superior | Middle | Posterior |
|---|---|---|
| Teratoma including dermoid cyst | Bronchogenic cyst | Neurogenic tumor |
| Pericardial cyst | Enterogenous cyst | |
| Normal thymus | Pulmonary sequestration | |
| Lymphoma | ||
| Vascular malformation | ||
| Thymic cyst | ||
| Cystic hygroma | ||
| Intrathoracic goiter |
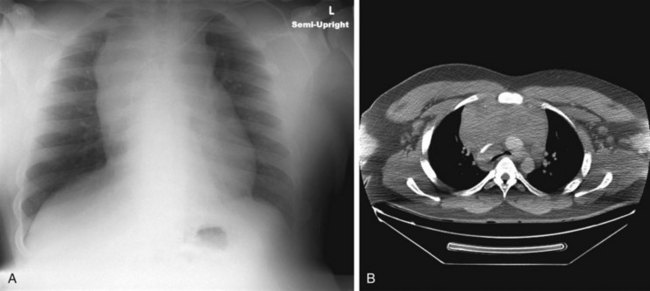
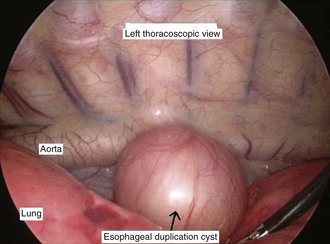
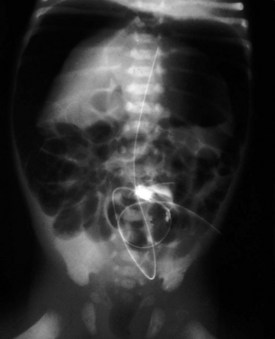
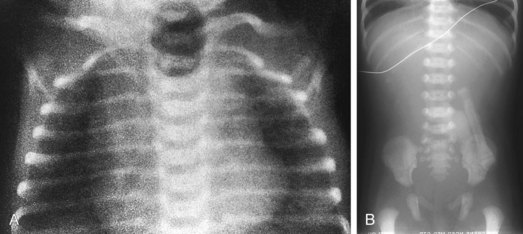
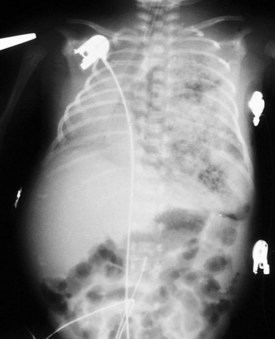
Esophageal atresia with or without tracheoesophageal fistula is a common cause of airway obstruction. The inability to pass a Replogle (nasogastric) tube into the stomach clarifies this diagnosis. This tube also serves as a sump catheter to drain the proximal pouch and to limit upper airway contamination associated with this anomaly. Infants develop respiratory distress secondary to esophageal obstruction and the tracheoesophageal communication. Neonates with esophageal atresia characteristically have excessive salivation and coughing due to the pooling of secretions in the proximal pharyngeal pouch. Most patients are diagnosed by their inability to tolerate their initial feedings. The diagnosis of congenital esophageal obstruction secondary to esophageal atresia is frequently made on antenatal ultrasonography by the presence of microgastria, polyhydramnios, and frequent fetal hiccups. Although several major variants of this condition exist, the most common, a blind proximal pouch with a distal tracheoesophageal fistula, occurs in approximately 85% of all patients. Inspired air from the trachea communicates directly to the stomach via a fistulous connection (Fig. 17-29). This leads to gastric distention and retrograde gastroesophageal reflux into the lungs, precipitating respiratory distress. Positive-pressure ventilation by either mask or endotracheal tube should be avoided in these patients before surgery because of the risk of gastric distention and perforation, severe retrograde reflux, and pneumonia (Fig. 17-30). The second most common variant of this condition is pure esophageal atresia, which on plain radiography shows a dilated proximal pouch and a gasless abdomen (Fig. 17-31). On physical examination these infants have a scaphoid abdomen due to the lack of distal air passage into the bowels (Fig. 17-32). Isolated tracheoesophageal fistula without esophageal atresia is the third most common form of this anomaly. These children lack esophageal obstruction and may at times be diagnosed at later ages with symptoms of persistent cough or recurrent pneumonia. Although called an H-fistula, the appearance is more N-shaped with a more proximal communication with the trachea and distal communication into the esophagus. The diagnosis of this variant is made more challenging because of this acute angle of communication between the esophagus and trachea, which inhibits reflux of orogastric contents into the airway (Fig. 17-33).
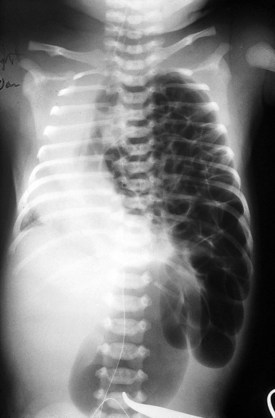
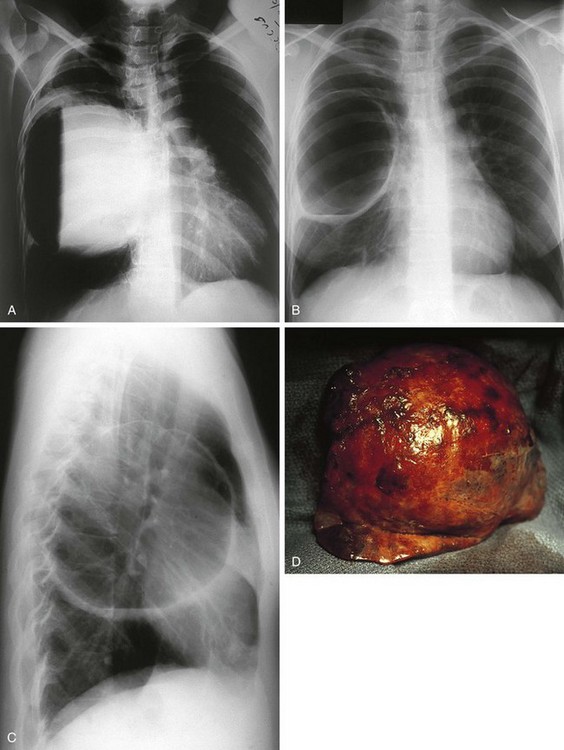
Congenital diaphragmatic hernia is a common cause of respiratory distress in newborns, occurring with an incidence of approximately 1 in every 4000 live births. Defects in the diaphragm may result either from abnormal fusion of the posterolateral pleuroperitoneal membrane (foramen of Bochdalek) or from defects in formation of the central diaphragmatic muscle (foramen of Morgagni hernia). Diffuse muscular weakness may give rise to diaphragmatic eventration or postcardiac surgical or birth injury to the phrenic nerve. Congenital diaphragmatic abnormalities of the foramen of Bochdalek are the most common form of lesion and are typically left sided in 85% of patients (Fig. 17-34). These defects occur early in gestation, allowing abdominal contents to herniate into the chest. This limits lung expansion and growth, displacing the heart, resulting in pulmonary hypoplasia and persistent pulmonary hypertension. Many of these infants have severe respiratory distress occurring shortly after umbilical cord division. On physical examination there is marked nasal flaring, chest wall asymmetry (larger contralateral hemithorax secondary to lung hyperplasia), displaced heart tones to the side opposite the hernia, and ipsilateral absence of breath sounds with dullness to percussion due to the presence of abdominal viscera in that hemithorax. Plain chest radiography demonstrates intestinal loops within the hemithorax displacing the cardiomediastinal silhouette to the opposite side. Infants with foramen of Bochdalek congenital diaphragmatic hernias have severe respiratory failure secondary to both pulmonary hypoplasia and pulmonary hypertension. Despite aggressive therapy with nitric oxide, high-frequency oscillatory ventilation, and extracorporeal membrane oxygenation (ECMO), mortality remains approximately 50% (Fig. 17-35).
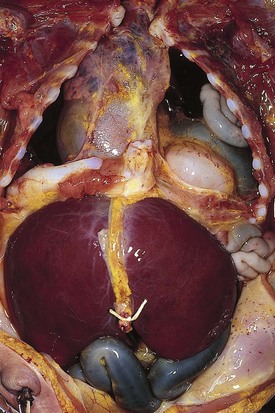
Diaphragmatic eventration may have a radiographic appearance similar to that of diaphragmatic hernia but usually lacks acute neonatal presentation. Foramen of Morgagni diaphragmatic hernias represent less than 5% of all diaphragmatic hernias. Often they are incidental findings seen on routine radiography obtained for other reasons (typically a cough). Morgagni hernias typically have a sac, which may include the transverse colon, liver, or small bowel (Fig. 17-36). Intestinal incarceration is a rare complication. Cystic pulmonary adenomatoid malformations may be confused with diaphragmatic hernia on plain films and are usually distinguished by the location of the gastric air bubble (Fig. 17-37).
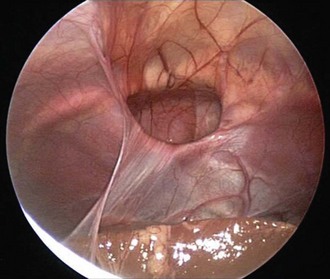
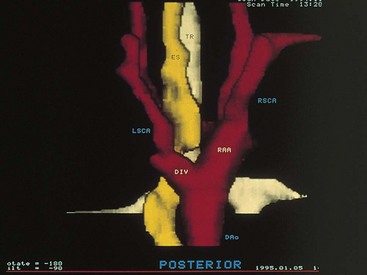
Vascular ring anomalies may be a source of tracheoesophageal compression giving rise to varying degrees of dyspnea or dysphagia. These anomalies originate from persistence of the embryonic aortic arches. Plain film findings demonstrating narrowing of the mediastinal portion of the tracheal air contour suggest the presence of a vascular ring. The diagnosis may be confirmed by nuclear magnetic resonance imaging (MRI), contrast barium swallow, or endoscopic evaluation of the airway. MRI has replaced aortography for delineating the associated vascular anatomy (Fig. 17-38).
Lung
Lung Bud Anomalies (Bronchopulmonary Foregut Malformations)
Four major bronchopulmonary foregut malformations exist: congenital lobar emphysema (CLE), congenital pulmonary adenomatoid malformation (CPAM), pulmonary sequestration, and bronchogenic cyst. The most common of these, CPAM, results from the proliferation of primordial bronchial structures in the absence of alveoli. CPAMs are subclassified as one of three variants on the basis of their size, shape, and pathologic appearance. Type I CPAM comprises single or multiple cysts greater than 2 cm in diameter and lined with ciliated pseudostratified columnar epithelium. Type I lesions may be difficult to distinguish from diaphragmatic hernias (Fig. 17-39). Type II lesions are small cysts, less than 1 cm in diameter, and lined with cuboidal to columnar epithelium. These lesions are associated with a broad spectrum of congenital anomalies. Type III CPAMs are large benign cysts lined with ciliated cuboidal epithelium or solid masses. These are often fatal and have a high incidence of associated anomalies.
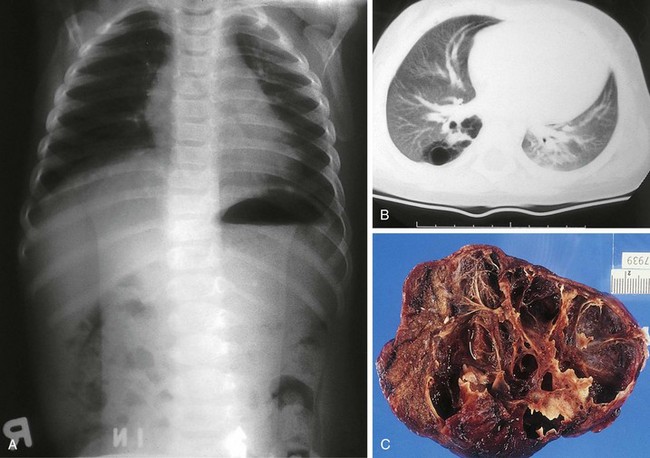
Figure 17-39 Macrocystic adenomatoid malformation seen on plain film (A), CT scan (B), and surgical specimen (C).
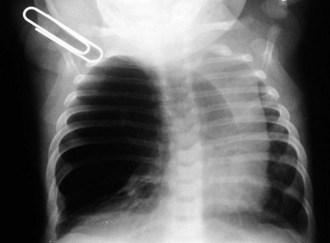
Congenital lobar emphysema is a condition that results from a segment of poorly developed or absent cartilage in the tracheobronchial tree and leads to lung hyperexpansion secondary to a “check valve effect” and air trapping. Subsequent lung overdistention may lead to respiratory distress, pneumonia, and mediastinal shift. Many of these patients present with symptomatic lesions in the first few weeks of life; however, other patients have a more indolent progression of symptoms over the first 6 months of life. Other patients may remain entirely asymptomatic. The plain radiographic appearance of these patients demonstrates lung hyperlucency and hyperexpansion in the upper or middle lobes (Fig. 17-40). Acute cardiopulmonary decompensation may occur in otherwise healthy patients with this anomaly, due to positive-pressure ventilation such as that which might occur at the time of the induction of general anesthesia for surgery (Fig. 17-41).
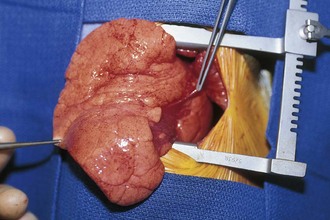
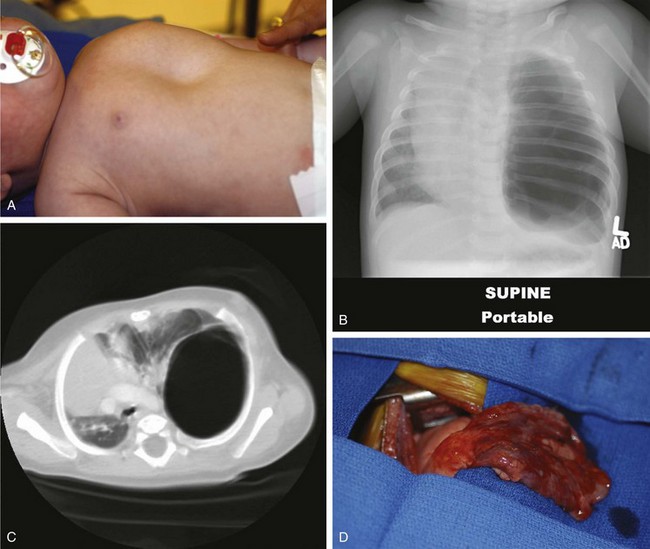
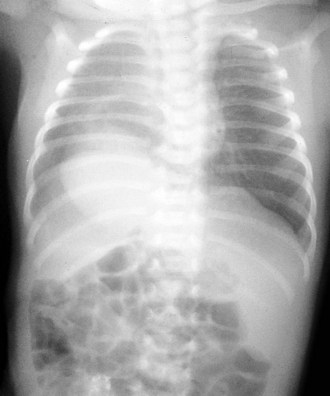
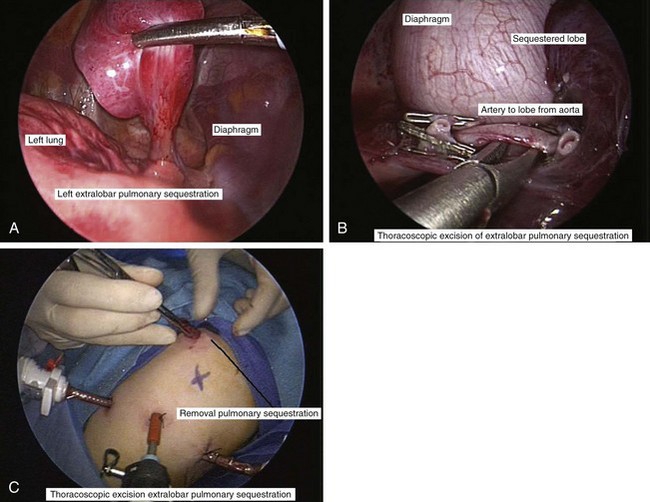
Primary pulmonary blastoma is a stromal malignancy of the lung that may present with unilateral hyperinflation, mimicking and possibly confused with lobar emphysema (Fig. 17-42). Pulmonary sequestrations are accessory pulmonary parenchymal tissue that lack direct tracheobronchial communication. These anomalies receive their blood supply from the systemic circulation. They may arise from within the pulmonary parenchyma (intralobar) (Fig. 17-43) or reside separately from normal lung tissue (extralobar) (Fig. 17-44). Although commonly found in the left costophrenic sulcus, sequestrations may be located in either hemithorax or in the abdomen. They may also communicate with other foregut structures in the gastrointestinal tract owing to their shared embryonic origin. Usually asymptomatic and found on routine chest x-ray or noted as an incidental finding during another thoracic procedure, these lesions may be a source of recurrent intrathoracic infection and should undergo elective resection. Duplex ultrasonography or, more commonly, CT or MRI evaluation may be used in the diagnostic assessment to demonstrate systemic arterial blood supply (Fig. 17-45).
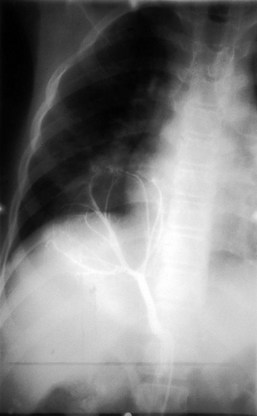
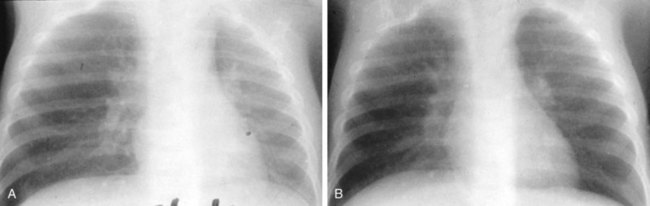
Pneumothorax may occur as a result of thoracic trauma, cystic fibrosis, or spontaneously (Fig. 17-46). Patients may develop acute severe pleuritic chest pain and associated dyspnea. Physical examination findings may demonstrate hyperresonance and diminished breath sounds over the ipsilateral hemithorax. Mediastinal shift, jugular venous distention, hypotension, and diaphragmatic flattening may result from the development of a tension pneumothorax. This requires emergency life-saving needle decompression followed by thoracostomy tube placement. A subpopulation of young patients, usually male and asthenic in build, may present with acute spontaneous pneumothorax. Spontaneous pneumothorax is typically secondary to apical bullous lung disease (Fig. 17-47). The etiology of this condition is unknown. These patients usually require chest decompression by thoracostomy tube placement. Recurrent episodes of spontaneous pneumothorax are an indication for surgical exploration with resection of the apical bullae and either mechanical or chemical pleurodesis.
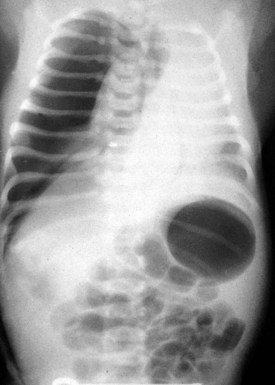
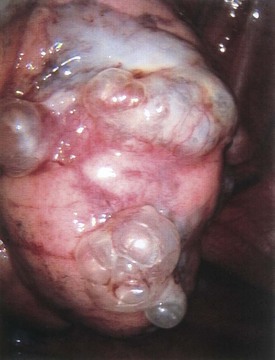
On occasion, surgical intervention may be necessary in the treatment of pulmonary infections that persist despite aggressive antibiotic therapy (Fig. 17-48). The development of an intrathoracic empyema as a sequela of streptococcal or staphylococcal pneumonia may restrict lung expansion. Surgical intervention may provide the means of diagnosis and the ability to rule out bronchial foreign body obstruction (Fig. 17-49); the capacity to assess for evidence of malignancy; or allow treatment through providing adequate drainage or mechanical pleural clearance. In addition, surgery may provide a means of treatment of chest lesions that may have become secondarily infected (Fig. 17-50). Thoracoscopy with video-assisted thoracic decortication may hasten recovery from pneumonia and these parapneumonic consequences. More recent experience supports the use of chest tube drainage with instillation of fibrolytic therapy (urokinase or tissue plasminogen activator) in the treatment of pediatric empyema.
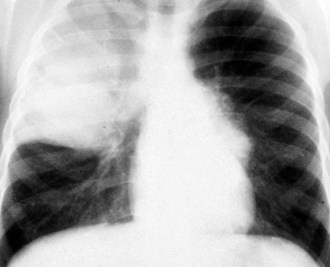
Chest Wall
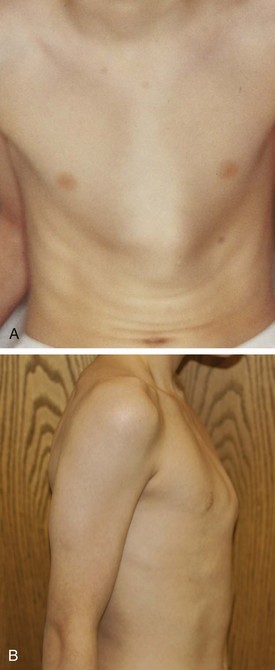
Pectus excavatum (“funnel chest”) is the most common congenital chest wall deformity (Fig. 17-51). This condition is characterized by the posterior angulation of the sternum toward the spine and abnormalities of the costal cartilages. Pectus excavatum has a 3 : 1 male predominance. Although often not impressive during infancy, this deformity increases during childhood and adolescence. This chest wall malformation typically causes no cardiopulmonary symptoms or disability. However, there is a subset of patients who will have exercise intolerance, mitral valve prolapse, or gastroesophageal reflux that may be attributable to this deformity. Some debate exists as to the relationship of these symptoms to the defect. Although the psychological implications of this deformity in teenagers relative to their self-esteem may seem to be a more compelling indication for surgical repair, there is evidence that in active patients, improved stamina is achieved after repair of severe pectus excavatum chest wall deformities. A minimally invasive technique, the Nuss procedure, has made the repair of pectus excavatum more appealing to many patients and their families.

Pectus carinatum (“pigeon chest”) is a protrusion deformity of the chest wall (Fig. 17-52). This condition represents a spectrum of sternal and midchest anomalies that may give rise to this malformation. Pectus carinatum occurs more commonly in males than females and is usually asymptomatic. Surgical correction of carinatum deformities has been replaced almost completely by nonsurgical chest bracing or compression devices, which are custom fitted and worn by patients for various time periods. Marfan syndrome must be considered in pectus carinatum or pectus excavatum deformities. The coexistence of other conditions such as aortic root abnormalities and ocular lens subluxation should be evaluated in these patients. Poland syndrome is a rare chest wall deformity that consists of a constellation of abnormalities including unilateral agenesis or dysplasia of the rib cage and chondral cartilages, absence of pectoralis major and minor muscles, hand deformities, and breast and areolar defects (Fig. 17-53). Other chest wall deformities include sternal cleft and pentalogy of Cantrell, discussed later (see Abdominal Wall Defects). Ectopia cordis is often complicated by the presence of severe congenital heart disease.
< div class='tao-gold-member'>
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


