Structural Abnormalities of the Genitourinary Tract
George W. Kaplan
Irene M. McAleer
A discussion of neonatal pediatric urology in essence encompasses most of pediatric urology. Although, there are age-specific problems that do not present in early infancy, problems present primarily or specifically in the neonatal period. It is these latter lesions on which this discussion will focus. The almost universal use of antenatal ultrasonography, especially with Doppler and 3-dimensional (3D) imaging, has had a profound effect on detection, management, and understanding of many lesions of the urinary tract. Genitourinary anomalies account for approximately 50% of all antenatally sonographically detected lesions; hydronephrosis represents about two-thirds of these genitourinary abnormalities (1). Information from ultrasonography has been further complemented with magnetic resonance imaging (MRI) imaging, fetal bladder urine specimen measurements of electrolytes, osmolality, and β2-microglobulin. In utero surgical therapy is also possible with improved fetal surgical techniques, although the benefits derived from fetal surgery are not clear (2,3,4,5).
To properly understand and interpret the symptoms and findings seen in most congenital urologic problems, understanding the significant events of embryogenesis of the lesions seen is essential. Abnormalities of embryogenesis will be addressed as the resulting anomalies are covered.
The ureteral bud arises from the mesonephric duct at 4 to 5 weeks of gestation, the kidney begins to form at 6 weeks, and the bladder develops during the sixth to seventh week. The Wolffian duct is then incorporated into the bladder (Fig. 43-1). It is not until week 10 of gestation that urine production begins, and it is usually not until 14 to 16 weeks of gestation that the urinary tract is evident on antenatal ultrasonography (6). A number of anomalies may result from disordered embryogenesis of the ureter or kidney. Most genitourinary anomalies seem to occur sporadically, although some of the abnormalities are familial and others are related to chromosomal abnormalities.
IMAGING
The overall incidence of detection of fetal anomalies is currently about 1%. Urologic diagnostic imaging in the fetus, the newborn, and, to some extent, in the infant, is limited by the renal function (see Fig. 43-1). Neonatal renal functional parameters may be sufficient to maintain homeostasis: they may not suffice to produce the accuracy of diagnosis as expected from some imaging examinations used in older children and adults.
Ultrasonography and voiding cystourethrography are not dependent on renal function and, hence, assume even greater diagnostic import than in other age groups. Improved ultrasonographic equipment, resolution, and techniques are identifying fetal anomalies with greater accuracy resembling similar studies obtained in children and adults. Doppler imaging of the renal vessels in utero can suggest the presence of 1 or 2 functioning kidneys and Doppler imaging of the hypogastric vessels can delineate the presence or absence of the bladder if there is difficulty in reliably locating the bladder. Intravenous urography and, to a lesser extent, renal scintigraphy depend on renal function to produce images and consequently may be less reliable than in the adult; newer scintigraphic agents and techniques have made evaluation of the neonatal kidneys more reproducible and reliable (Fig. 43-2).
ANOMALIES OF THE KIDNEY
With normal embryogenesis as background, the anomalies and problems that may be encountered can be anticipated, because most have an embryologic basis. Some are incompatible with life, which may lead to an early demise; some are of no clinical significance, but most are potential sources of morbidity that can be minimized if the children are treated at an early age.
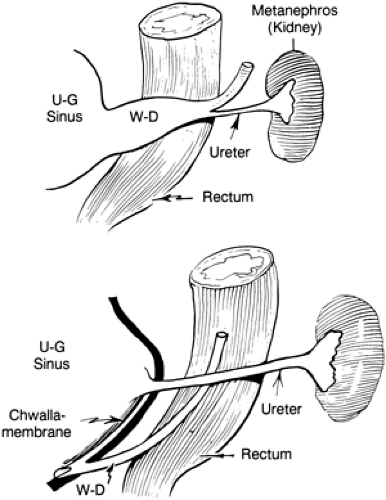 Figure 43-1 Incorporation of the wolffian (i.e., mesonephric) duct (W-D) into the urogenital sinus (U-G). From Kelalis PP, King LR, Belman AB, eds. Clinical pediatric urology, vol. 1. Philadelphia: WB Saunders, 1976:504, with permission. |
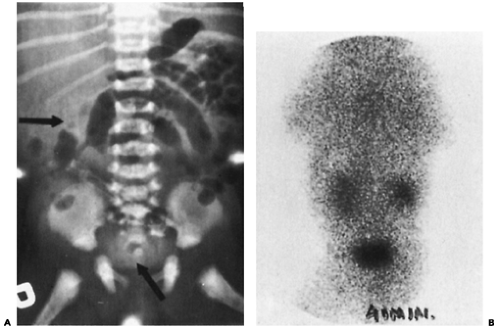 Figure 43-2 A: An excretory urogram in a newborn infant. The right kidney barely concentrates contrast (arrow); there is no evidence of the left kidney. Contrast is apparent in the bladder (arrow). B: An isotope renal scan the next day demonstrates the presence and function of two kidneys. The child was normal. |
Renal anomalies include the cystic diseases and abnormalities of number, position, and rotation. If the pronephros fails to develop, the mesonephros will not develop, resulting in renal and ureteral agenesis and absence of the ipsilateral genital ducts in the male. If the mesonephric duct develops but the mesonephros does not, there will be renal agenesis but the genital ducts will be present and there might be a blind-ending ureter.
Nephrogenesis is induced by the ureter; therefore, normal nephrogenesis depends on the ureteral bud meeting normal metanephrogenic blastema (7). If the ureter fails to meet the metanephric blastema, a blind-ending ureter might result. When the ureter meets a degenerating portion of the metanephrogenic blastema (i.e., the cephalic or caudal end of the blastema), renal dysplasia may result (7).
Renal Agenesis
Renal agenesis can be unilateral or bilateral. Bilateral renal agenesis obviously is incompatible with extrauterine life. In cases of bilateral renal agenesis, there is no in utero urine production; thus, there is marked oligohydramnios and the affected fetus may exhibit the deformational changes of Potter syndrome (8). Adequate amniotic fluid is required for normal lung development so the lungs may be hypoplastic. The incidence of bilateral renal agenesis is roughly 1 in
4,000 to 5,000 births (8). Renal agenesis sometimes may be associated with sirenomelia (8). Bilateral renal agenesis can be diagnosed antenatally by a combination of sonographic findings that include lack of identifiable renal masses, no demonstrable renal vessels on Doppler imaging, absent bladder, and severe oligohydramnios (6). For reasons that are unclear, some infants with this diagnosis have normal or near-normal levels of amniotic fluid. Renal agenesis must be considered postnatally when Potter facies are noted, when there is no urinary output within 24 to 48 hours, or when there is evidence of ventilatory failure with small lungs on chest radiograph. The postnatal diagnosis can be inferred ultrasonographically by the absence of identifiable kidneys or renal vessels with Doppler and the absence of urine in the bladder. Renal scintigraphy is used to prove that there is no identifiable functioning renal tissue. When the postnatal diagnosis of bilateral renal agenesis is made and confirmed, attempts at life support, which may have been initiated because of respiratory distress, should, in our opinion, be abandoned. There may be a familial inheritance of bilateral renal agenesis, with one review reporting up to 3% of siblings of probands having bilateral renal agenesis (9).
4,000 to 5,000 births (8). Renal agenesis sometimes may be associated with sirenomelia (8). Bilateral renal agenesis can be diagnosed antenatally by a combination of sonographic findings that include lack of identifiable renal masses, no demonstrable renal vessels on Doppler imaging, absent bladder, and severe oligohydramnios (6). For reasons that are unclear, some infants with this diagnosis have normal or near-normal levels of amniotic fluid. Renal agenesis must be considered postnatally when Potter facies are noted, when there is no urinary output within 24 to 48 hours, or when there is evidence of ventilatory failure with small lungs on chest radiograph. The postnatal diagnosis can be inferred ultrasonographically by the absence of identifiable kidneys or renal vessels with Doppler and the absence of urine in the bladder. Renal scintigraphy is used to prove that there is no identifiable functioning renal tissue. When the postnatal diagnosis of bilateral renal agenesis is made and confirmed, attempts at life support, which may have been initiated because of respiratory distress, should, in our opinion, be abandoned. There may be a familial inheritance of bilateral renal agenesis, with one review reporting up to 3% of siblings of probands having bilateral renal agenesis (9).
The incidence of unilateral renal agenesis is roughly 1 in 500 to 1,500 (8,10). There is a higher incidence of contralateral renal abnormality in patients with unilateral renal agenesis when compared to the general population, being generally obstructive or refluxing in nature. One-third of the patients with a solitary kidney in one series required some form of surgical procedure on the solitary unit (11). Unilateral renal agenesis may be associated with congenital scoliosis and with vaginal and uterine agenesis (12,13). It is the most common nonskeletal anomaly seen with imperforate anus (14). Unilateral renal agenesis previously was not thought to affect longevity or health as long as the contralateral kidney was normal. Recent experimental studies have suggested that renal injury may be produced in the solitary kidney by hyperfiltration and that a reduced protein diet may be somewhat protective. Clinical studies of patients with a solitary kidney (e.g., transplant donors, trauma victims) have, at this point, failed to confirm this (15).
Brenner and associates have noted that most people with unilateral renal agenesis do not develop progressive renal disease but longstanding hyperfiltration in most people with a solitary kidney from any etiology may cause glomerular hypertension which may be the culprit for developing progressive glomerular damage with subsequent proteinuria and renal insufficiency (16). Goldfarb and associates reviewed renal function in donor nephrectomy patients and found that renal function was well preserved in most donors over a prolonged follow-up averaging 25 years with some increase in proteinuria found in some patients but with only marginal significance in the group as a whole (17).
Renal Ectopia and Fusion
Failure of renal ascent will result in a pelvic kidney and may be associated with vaginal or vertebral anomalies (12). If the two metanephrogenic masses come into contact with each other in the pelvis, they may fuse and form a pancake or a horseshoe kidney (13). The embryogenesis of crossed ectopia, with or without fusion, is harder to explain but might result from lateral bending and rotation of the tail bud of the embryo, thereby altering the course of ascent (13).
Renal malrotation, or incomplete rotation, occurs when the ascending kidney, maintains its early fetal orientation and its renal pelvis is directed anteriorly. Malrotation is routinely present in fusion anomalies and in pelvic and crossed ectopias, and is also occasionally seen in kidneys located in the renal fossa. Incomplete rotation is of no clinical significance, but may cause difficulties in some imaging study interpretation: it may also be important in planning reconstructive procedures.
Abnormalities of renal position (i.e., ectopia) are interesting anomalies that generally, are of no clinical import but may become apparent because of trauma (i.e., hematuria), a palpable mass, or associated urologic abnormality. The ectopic kidney may be located in the chest or the pelvis. Thoracic kidneys usually are associated with eventration of the diaphragm and are of no clinical significance except as a finding on a chest radiograph (18). Pelvic ectopia is the most common of the abnormalities of position and often is associated with vesicoureteral reflux or ureteropelvic junction obstruction (19). Additionally, girls with müllerian anomalies have an increased incidence of pelvic kidney compared to the general population; hence, the finding of a pelvic kidney in a girl warrants further investigation of the genital tract to uncover associated anomalies (20).
Fusion anomalies, such as horseshoe or pancake kidneys, may present in the same way that anomalies of position present. Horseshoe kidneys are found with increased frequency in girls with Turner syndrome (21). There is an increased incidence of ureteropelvic junction obstruction in horseshoe kidneys (19). Some patients with horseshoe kidneys may have increased stone formation, as a result of relative urinary stasis with drainage from the nondependent renal pelvis. Crossed ectopia with or without fusion (i.e., one kidney is found on the opposite side of the side of ureteral bud development) is uncommon. When crossed ectopia occurs, the left kidney more commonly crosses to the right side than vice versa (22). By definition, the ipsilateral ureteral orifice is located on the anatomically appropriate side of the body (i.e., the left ureter is on the left side of the trigone and the right ureter is on the right side of the trigone). There is an increased incidence of vesicoureteral reflux and of ureteropelvic junction obstruction in crossed ectopic kidneys (19). Patients with crossed ectopia have an increased incidence of skeletal and cardiac abnormalities (14).
Supernumerary Kidney
The presence of a supernumerary (i.e., third) renal mass is a very rare anomaly; the clinical significance of this is determined by associated pathologic conditions (23). The supernumerary kidney usually is small, and more often caudal than cranial to the normally placed kidney. Many patients and some physicians confuse a supernumerary
kidney with duplication of the collecting system and incorrectly, refer to ureteral duplication as a third kidney.
kidney with duplication of the collecting system and incorrectly, refer to ureteral duplication as a third kidney.
Cystic Disease
Renal cystic diseases are a group of disorders seen in pediatric urologic practice that frequently will present in the neonatal period. Because there is no uniform system of classification, there can be difficulty in communication between disciplines. An accurate diagnosis is needed for prognosis and for genetic counseling. It is for this reason that communication must be clear. Table 43-1 is a classification scheme that has been clinical useful in our practice.
Autosomal recessive polycystic kidney disease (ARPCKD), as the name implies, is an inherited disorder whose mode of transmission follows a mendelian recessive pattern. Its reported incidence is between 1 in 6,000 to 1 in 14,000 pregnancies. In this disorder, formerly known as infantile polycystic disease, the kidneys are very large and often occupy the entire retroperitoneum (Fig. 43-3). The cysts are small and are actually enlargements of the collecting ducts (24). The liver almost always is abnormal. At times there will be periportal hepatic fibrosis as a significant part of this complex. Death in the neonatal period is secondary to either renal or pulmonary failure. Those who survive the neonatal period usually will exhibit decreased renal function and hypertension, but at times liver failure as a result of hepatic fibrosis may be the most prominent part of the clinical picture (25). Imaging studies such as antenatal or postnatal ultrasonography, computed tomography (CT) or intravenous urography usually are diagnostic with very large kidneys demonstrating a sunburst streaking pattern.
ADPCKD is inherited in a mendelian dominant fashion and is more common than the recessive form. It usually does not present until adult life, hence its former name, adult polycystic disease. These patients usually present with hypertension, hematuria, urinary tract infection, or renal failure. When this problem presents in childhood, it may do so as an abdominal mass or may be found with ultrasound as either antenatal evaluation, screening for polycystic disease, or coincidentally when an ultrasound is being obtained for some other evaluation. Imaging studies will prove diagnostic, because multiple large cysts that splay and distort the collecting system will be present. Although there often are associated hepatic cysts, liver failure is not usually a clinical feature of this disorder. Microdissection studies have shown that the cysts are as a result of abnormal branching of the collecting tubules and cystic dilations of portions of the nephron (26). Generally, detectable cysts may not develop until middle to late adult life; hence, the disease is undetectable clinically until they appear.
TABLE 43-1 CLASSIFICATION OF RENAL CYSTIC DISEASE | |
|---|---|
|
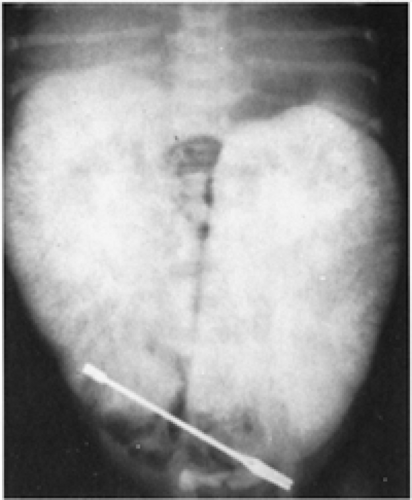 Figure 43-3 An excretory urogram in a 2-day-old girl with autosomal recessive polycystic kidney disease shows the sunray appearance of the contrast material and the enormous renal size. From Kelalis PP, King LR, Belman AB, eds. Clinical pediatric urology, vol. 2. Philadelphia: WB Saunders, 1976:686, with permission. |
Tuberous sclerosis can mimic both autosomal recessive and autosomal dominant polycystic disease in that lesions grossly similar to either form of polycystic disease can be found in the tuberous sclerosis complex (27). Microscopically, lesions characteristic of tuberous sclerosis will be seen on biopsy of the affected kidneys. Angiomyolipomas (i.e., renal hamartomas), are the more commonly seen renal lesions in patients with tuberous sclerosis.
Multicystic kidney disease, with a frequency of 1 in 3,000 pregnancies (11), is the most common form of cystic
disease seen in neonates. Originally defined by Spence and associates (28), this is a unilateral (or bilateral) lesion in which the entire kidney is replaced by cysts of varying sizes. Grossly, there is no recognizable renal tissue present, but microscopically there may be dysplastic renal elements in the septa between the cysts (Fig. 43-4). Bilateral multicystic kidney disease is incompatible with life. Multicystic kidney disease is sporadic and is not inherited. Some multicystic kidneys involute, probably by absorption of the cyst fluid. This can occur antenatally or in the first few months of life. It is likely that many cases of presumed renal agenesis are multicystic kidneys that have undergone involution.
Multicystic kidneys usually are detectable with antenatal ultrasonography, but occasionally can present during infancy as palpable masses. Ultrasonography will demonstrate multiple, variably sized cysts in a random pattern (Fig. 43-5), and the affected kidney is generally without function on renal scan or intravenous urography.
disease seen in neonates. Originally defined by Spence and associates (28), this is a unilateral (or bilateral) lesion in which the entire kidney is replaced by cysts of varying sizes. Grossly, there is no recognizable renal tissue present, but microscopically there may be dysplastic renal elements in the septa between the cysts (Fig. 43-4). Bilateral multicystic kidney disease is incompatible with life. Multicystic kidney disease is sporadic and is not inherited. Some multicystic kidneys involute, probably by absorption of the cyst fluid. This can occur antenatally or in the first few months of life. It is likely that many cases of presumed renal agenesis are multicystic kidneys that have undergone involution.
Multicystic kidneys usually are detectable with antenatal ultrasonography, but occasionally can present during infancy as palpable masses. Ultrasonography will demonstrate multiple, variably sized cysts in a random pattern (Fig. 43-5), and the affected kidney is generally without function on renal scan or intravenous urography.
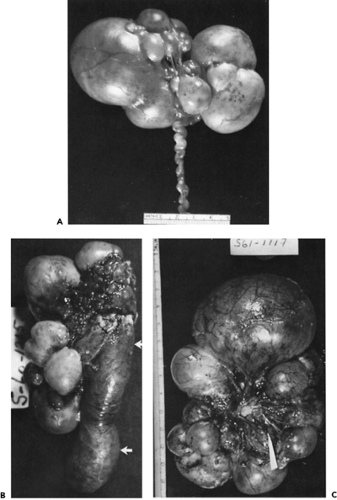 Figure 43-4 Three examples of the variety of multicystic diseases. A: Multicystic kidney with a torturous atretic ureter. The cysts vary in size and appear to be held together by fibrous tissue. B: Multicystic kidney in a 1-month-old girl. The dilated pelvis and proximal ureter are indicated (arrows). C: Multicystic kidney in a 4-day-old girl. No ureter was found during the nephrectomy. From Kelalis PP, King LR, Belman AB, eds. Clinical pediatric urology, vol. 2. Philadelphia: WB Saunders, 1976:210, with permission. |
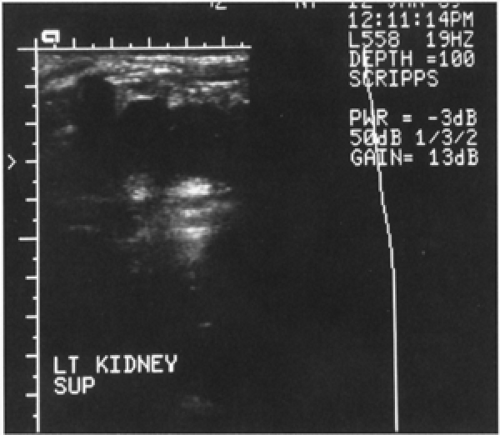 Figure 43-5 An abdominal ultrasonographic study carried out in an infant with multicystic (i.e., cystic dysplastic) kidney. |
There is a great deal of controversy surrounding the management of these lesions; traditional therapy had been nephrectomy, but many pediatric urologists advocate observation because the incidence of sequelae such as infection, pain, hypertension, or malignancy is very low. Nephrectomy seems a reasonable alternative to lifelong follow-up, however, and is our current recommendation if the kidney does not completely involute within 6 to 12 months. At the present time, a Multicystic Kidney Registry is collecting data on cases that are followed with observation. Roughly 25% of the patients with multicystic kidney disease have an obstructive lesion such as ureteropelvic junction obstruction on the contralateral side, and it is this fact that will determine the patient’s ultimate prognosis. Since the inception of the Multicystic Kidney Registry, vesicoureteral reflux has been found to be more common than obstruction. Involution, if it occurs, may take many years to occur; up to 50% of those followed for 3 to 5 years had no change in the multicystic kidney appearance on ultrasound (29).
ANOMALIES OF THE URETERS AND BLADDER
Duplication and Triplication of the Ureters
Multiple ureteral buds or premature division of the ureteral bud could produce ureteral duplication or triplication (30). If there are multiple ureteral buds, one bud is likely to meet degenerating rather than normal nephrogenic tissue. This could account for the increased incidence of renal dysplasia in the upper pole of a duplicated system (4). Duplication of the urinary collecting system is one of the more common abnormalities seen in the urinary tract; its occurrence is about 0.8% (30). Approximately 12% of sibs and parents were affected with ureteral duplication when reviewing family inheritance probands. (9) Duplication can be either complete or incomplete. Incomplete duplication is usually of no clinical significance, there can be ureteroureteral reflux between the two limbs of the partial duplication resulting in dilation of one of the ureters, usually the lower one. Complete duplication occurs once in every 500 cases (30). Complete duplication, generally of no clinical significance, is associated with a higher incidence of other abnormalities in the urinary system. These abnormalities include both vesicoureteral reflux and obstruction.
Vesicoureteral reflux probably is the more common of the anomalies associated with ureteral duplication, and usually occurs into the lower moiety of a duplicated system (Fig. 43-6). Duplication is seen in approximately one in five people with vesicoureteral reflux, which is much higher than its incidence in the general population (31). The grade of reflux associated with a complete duplication usually is greater than that seen with a single system. Obstruction is common when the upper moiety of a complete duplication is abnormal, Both obstruction and vesicoureteral reflux associated with duplications may present as either mass lesions or urosepsis; many of these duplications are diagnosed in utero, with hydronephrosis seen in the upper or lower segments of the duplicated systems.
If the ureteral bud arises from a locus that is more cranial or caudal than normal, ureteral ectopia, vesicoureteral reflux, or paraureteral diverticula might be produced (32). Ectopic ureteroceles probably result from abnormalities of the ureteral bud and ureteral ectopia (33). Simple ureteroceles are thought to be produced by persistence of the Chwalla membrane (i.e., the membrane covering the distal end of the ureter during development) (34).
Ureteral obstruction occurs primarily at the ureteropelvic and ureterovesical junctions. These obstructions usually are intrinsic in nature, and the ureter may be of normal or reduced caliber externally (35,36). Multicystic kidney disease has been said to result from ureteral obstruction early in gestation or may be secondary to disordered induction of the metanephrogenic mass by a faulty ureteral bud (37). Kitagawa and associates demonstrated an animal model for development of a multicystic kidney by obstructing the fetal lamb urethra (male) or ureter (female) at 60 days gestation, and proposed that the cysts developed only when the obstruction occurs after the glomeruli start to produce urine. (38)
Bladder Anomalies
Agenesis of the bladder could result if the allantoic stalk failed to develop (39); it also could occur if there was bilateral failure of ureteral migration with bilateral ureteral
ectopia, because migration of the ureters is necessary for formation of the trigone, which may be necessary for enlargement of the allantoic stalk (40). Urachal anomalies occur because of a general mesodermal failure, as in the prune-belly syndrome, or because of delayed closure of the urachus (41). Duplications of the bladder and urethra often are associated with duplications of the hindgut and lower spinal cord. Hence, it would seem that splitting of the hind end of the embryo might be responsible for this type of anomaly (42).
ectopia, because migration of the ureters is necessary for formation of the trigone, which may be necessary for enlargement of the allantoic stalk (40). Urachal anomalies occur because of a general mesodermal failure, as in the prune-belly syndrome, or because of delayed closure of the urachus (41). Duplications of the bladder and urethra often are associated with duplications of the hindgut and lower spinal cord. Hence, it would seem that splitting of the hind end of the embryo might be responsible for this type of anomaly (42).
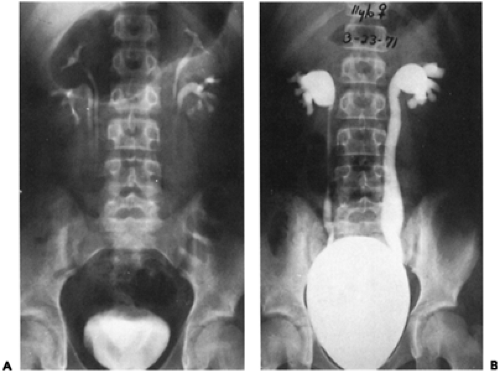 Figure 43-6 A: An intravenous urogram in a girl with complete, bilateral duplication of the collecting system. Note the blunting of the lower calyces. B: A cystogram in the same child demonstrates reflux into both lower collecting systems only. From Belman AB. The clinical significance of vesicoureteral reflux. Pediatr Clin North Am 1976;23:707, with permission. |
Posterior urethral valves probably result from abnormal insertion and persistence of the mesonephric ducts distal to the Müllerian tubercle (type I), or from persistence of the cloacal membrane (type III) (43). Type II valves probably do not exist as an obstructing lesion (see Fig. 43-17).
Ureteral Ectopia
Ureteral ectopia exists when the ureter opens in a position other than its normal location at the corner of the trigone. Ectopia may occur in ureters of single or duplex kidneys (Fig. 43-7). The most common form of ureteral ectopia is lateral ureteral ectopia, in which the ureteral orifice lies within the bladder lateral to its normal position. This is the etiologic mechanism for primary vesicoureteral reflux (see Vesicoureteral Reflux). Significant medial or distal ureteral ectopia, less common than lateral ureteral ectopia, will cause clinical pathologic conditions that vary depending on the location of the ureteral orifice and the gender of the patient. An abnormal proximal budding locus on the mesonephric duct allows the ureteral bud to remain in prolonged contact with the Wolffian duct, so that the medially ectopic ureteral orifice may open anywhere along the course of the Wolffian duct (Fig. 43-8). In males, this includes the posterior urethra, seminal vesicles, vas deferens, or epididymis (44). In females, the ectopic ureter may open into the urethra, the uterus, or proximal vagina, or along the course of Gartner’s duct in the anterolateral wall of the vagina. If a medially ectopic ureter opens within the confines of the bladder, no clinical abnormality occurs. If, however, the ureter opens within the confines of the bladder neck mechanism, obstruction of the involved renal unit or vesicoureteral reflux may occur.
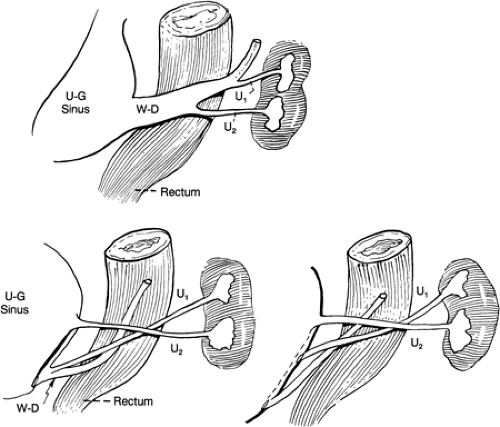 Figure 43-7 Development of ectopic ureter. U, ureter; U-G, urogenital; W-D, Wolffian duct. From Kelalis PP, King LR, Belman AB, eds. Clinical pediatric urology, vol. 1. Philadelphia: WB Saunders, 1976:510, with permission. |
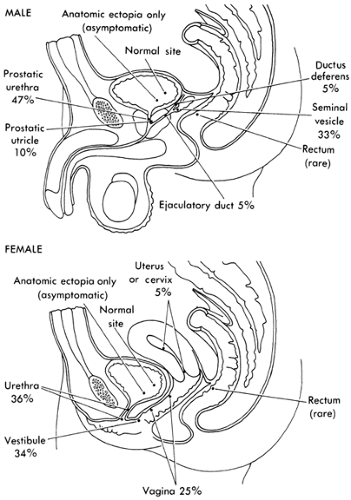 Figure 43-8 Sites of ectopic ureteral orifices and their relative frequencies of occurrence in men and women. From Gray SW, Skandalakis JE. Embryology for surgeons. Philadelphia: WB Saunders, 1972:536, with permission. |
In females, ectopic ureteral orifices that lie distal to the internal sphincter mechanism of the bladder neck can cause incontinence (45). Older girls usually present with constant dampness that is associated with an otherwise symptom-free, normal voiding pattern. Infant girls may be constantly wet or have a purulent discharge if the system is infected. Physical examination suggests the diagnosis if urine can be seen welling up in the vagina or if a spurt of urine is seen coming from the perineal ectopic ureteral orifice. Many are not diagnosed by physical findings alone. Eighty percent of these ectopic ureters arise from the upper pole segment of a total ureteral duplication, and CT, MRI, or diuretic renography with radionuclide agents will better delineate the anatomy. Occasionally, intravenous urography may suggest the diagnosis (45). However, the ectopic segment frequently functions poorly and it may not be visible on excretory urography even with delayed radiographs. A high index of suspicion and an awareness of the radiographic clues to a nonvisible duplication (i.e., the drooping lily sign) often will lead to the diagnosis (Fig. 43-9). Because an ectopic vaginal ureter may drain a poorly functioning and thus nonvisualizable single renal unit, congenital absence of one kidney in a girl with incontinence must not be accepted as a diagnosis without a thorough investigation. This investigation should include abdominal sonography, nuclear renal scan; occasionally computed tomography or MRI may be the only way to define the anatomy (46).
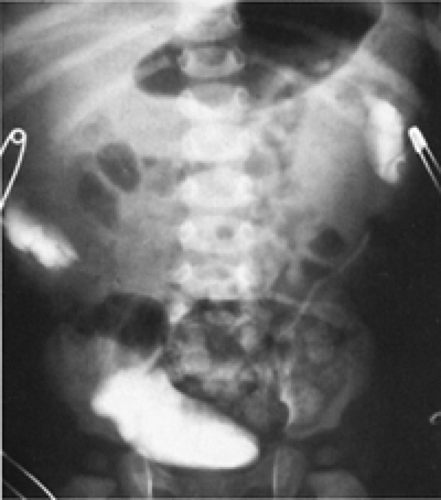 Figure 43-9 Excretory urogram in an infant with ureteral duplication. Ectopic ureters from upper segments produced massive displacement of lower segments, leading to a misdiagnosis of abdominal mass and neuroblastoma. From Kelalis PP, King LR, Belman AB, eds. Clinical pediatric urology, vol. 1. Philadelphia: WB Saunders, 1976:518, with permission. |
Treatment of the ectopic ureter depends on the presence or absence of significant function in the involved renal unit. If the ureter drains an otherwise healthy system, ureteral reimplantation into the bladder will correct the problem and preserve maximal renal function. If the ureteral anomaly is associated with a duplex kidney and there is good function in both segments of the kidney, ipsilateral ureteroureterostomy is indicated. Generally, the involved renal unit functions poorly, so excision of the involved segment is indicated. The distal ureteral stump is left undisturbed to avoid compromise of the normal sphincter mechanisms. The male infant with an ectopic ureter frequently presents with an antenatal ultrasound finding suggestive of ureteral dilatation, a mass, urinary tract infection, or epididymitis (44). In boys, an ectopic ureter will arise more frequently from a nonduplicated kidney, and can drain into the male genital tract anywhere from the prostatic urethra to the epididymis. Treatment is similar to that for females.
Ureterocele
A ureterocele is a cystic dilation of the distal submucosal or intravesical portion of a ureter. Ureteroceles account for a
broad spectrum of associated or secondary pathologic conditions, and constitute one of the more complex and confusing groups of anomalies of the lower urinary tract (47).
broad spectrum of associated or secondary pathologic conditions, and constitute one of the more complex and confusing groups of anomalies of the lower urinary tract (47).
Ureteroceles in children most commonly involve the end of the upper pole ureter of a duplex kidney (i.e., ectopic ureterocele), but may less commonly involve a single-system ureter (i.e., simple ureterocele) (48). The etiology of ureteroceles is uncertain. Failure of reabsorption of the Chwalla membrane from over the ureteral orifice has been proposed as an obstructive etiology (34). It seems more likely that ureteroceles result from an intrinsic defect in the ureteral bud itself, and from faulty or delayed incorporation of the ureteral bud into the urethra and bladder base (45).
Ureteroceles associated with a single-system ureter (i.e., simple ureteroceles) tend to be intravesical and in the normal position. Intravesical ureteroceles in children often are also associated with hydronephrosis of varying degrees (49).
Ureteroceles may be associated with significant derangement of the upper and lower urinary tract. The ureterocele most commonly originating from the upper pole ureter of a duplex kidney is associated with secondary pathology; the most frequently noted associated condition is hydronephrosis, with impaired function or dysplasia of the upper pole system and obstruction or reflux in the ipsilateral lower pole system (Fig. 43-10). Contralateral reflux or obstruction also may occur. The pathophysiology of the associated findings is easily understood by recognizing that a ureterocele may dissect under the trigonal epithelium and deform the ipsilateral or contralateral ureterovesical junction, causing various combinations of vesicoureteral reflux or obstruction in any or all of the ureters (50). Ten percent of ureteroceles occur bilaterally (47). If the ureterocele prolapses into, or otherwise occludes, the bladder outlet, bilateral hydronephrosis and relative bladder outlet obstruction may occur.
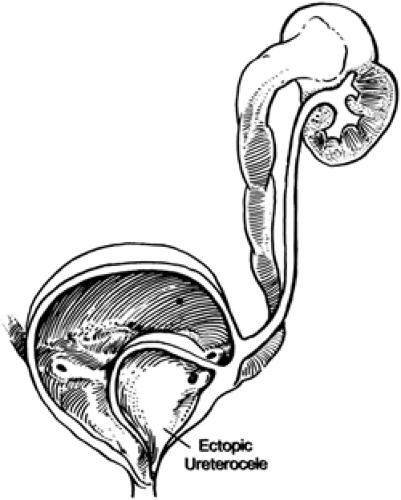 Figure 43-10 Ectopic ureterocele. From Malek RS, Kelalis PP, Burke EC, et al. Simple and ectopic ureterocele in infancy and childhood. Surg Gynecol Obstet 1972;134:611, with permission. |
Ureteroceles occur more commonly in females and usually manifest in early childhood; the ratio of occurrence in males and females is 1:6 cases (47). Most of our patients have presented during the first year of life. Most are now diagnosed antenatally with ultrasound but, historically, the most common presentation was that of a febrile infant found to have a urinary tract infection. If the ureterocele prolapses into the urethra, difficult voiding or azotemia may prompt evaluation. Ureterocele is the most common cause of urinary retention in the female infant. Rarely, a ureterocele will prolapse through the external urethral meatus in a female and present as an introital mass.
Classically, the diagnosis of a ureterocele is fairly straightforward. Renal and bladder ultrasonography will reveal the upper tract dilation and the wall of the ureterocele in the bladder (51). This can be seen antenatally. The intravenous urogram, if obtained, commonly shows complete ureteral duplication with upper pole hydronephrosis and the characteristic lucency of the ureterocele in the bladder. Cystography is necessary to establish the presence or absence of associated vesicoureteral reflux and to assess the integrity of the detrusor muscle backing the ureterocele. A diuretic renal scan can outline the function of the involved upper pole segment and any measurable obstruction of any of the segments, and is helpful in determining the best surgical approach.
The choice of treatment of a ureterocele depends on several factors. The age and clinical condition of the patient, the presence or absence of significant function in the involved renoureteral unit, and the presence of reflux or obstruction in the ipsilateral or contralateral uninvolved ureters all influence the choice of therapy. In the critically ill, septic infant, transurethral or transvesical unroofing or puncture of the ureterocele may provide decompression and allow stabilization of the child until his or her clinical condition allows definitive treatment. Alternatively, placement of a temporary percutaneous nephrostomy into the involved renal unit often can be done without the need for general anesthesia. The best form of definitive treatment has been debated over the years. If an intravesical ureterocele is associated with a single kidney and minimal hydronephrosis, simple excision of the ureterocele and reimplantation of the involved ureter should suffice. However, cystoscopic puncture of the ureterocele may also correct the problem without need for further surgical intervention (52). Even if the system is duplex, an en bloc ureteral reimplantation can be performed if the ureters are not too dilated (47).
Generally, definitive management of the hydronephrotic upper tract associated with a ureterocele depends on the function of the obstructed segment. If sufficient function exists in the involved unit, pyeloureterostomy or ureteroureterostomy to the ipsilateral uninvolved unit is
appropriate (47). In the most common situation, however, function usually is so poor that removal of the involved upper pole unit may be necessary (47). Some patients have had. distal ureterocele reconstruction without heminephrectomy with good initial results. The upper pole should be followed for resolution of the hydronephrosis. Some centers have described definitive endoscopic treatment of the ureterocele by puncturing the ureterocele with an electrocautery probe (53). Initially promising success rates have not proven to be as good as predicted (52). Woodard found that 80% of his patients required further surgery when transurethral puncturing of ureteroceles was used as primary therapy (178). Puncture of the ureterocele is appropriate initial management for unstable or septic patients and in select patients with intravesical ureteroceles as potentially definitive treatment. Debate centers over the management of the distal ureter and ureterocele itself. If upper pole nephrectomy has been performed, the distal ureter and ureterocele may collapse, alleviating any associated pathologic condition caused by the mass effect of the ureterocele. If there is significant reflux into the ureter associated with the ureterocele (which is uncommon), or if the ureterocele tends to evert into the dilated ureter prior to initial surgical intervention, excision of the involved ureter and marsupialization of the ureterocele is preferred. Depending on the function of the upper pole moiety, either heminephrectomy or upper to lower pole ureteroureterostomy generally is also necessary. This obviates the need for a secondary, delayed surgical excision of the ureterocele because of recurrent urinary tract infections or persistent reflux (50). At times, preliminary cutaneous diversion of the involved ureter with delayed reconstruction is warranted, to allow maximal recovery of function before deciding on the need for nephrectomy vs. reconstruction (47).
appropriate (47). In the most common situation, however, function usually is so poor that removal of the involved upper pole unit may be necessary (47). Some patients have had. distal ureterocele reconstruction without heminephrectomy with good initial results. The upper pole should be followed for resolution of the hydronephrosis. Some centers have described definitive endoscopic treatment of the ureterocele by puncturing the ureterocele with an electrocautery probe (53). Initially promising success rates have not proven to be as good as predicted (52). Woodard found that 80% of his patients required further surgery when transurethral puncturing of ureteroceles was used as primary therapy (178). Puncture of the ureterocele is appropriate initial management for unstable or septic patients and in select patients with intravesical ureteroceles as potentially definitive treatment. Debate centers over the management of the distal ureter and ureterocele itself. If upper pole nephrectomy has been performed, the distal ureter and ureterocele may collapse, alleviating any associated pathologic condition caused by the mass effect of the ureterocele. If there is significant reflux into the ureter associated with the ureterocele (which is uncommon), or if the ureterocele tends to evert into the dilated ureter prior to initial surgical intervention, excision of the involved ureter and marsupialization of the ureterocele is preferred. Depending on the function of the upper pole moiety, either heminephrectomy or upper to lower pole ureteroureterostomy generally is also necessary. This obviates the need for a secondary, delayed surgical excision of the ureterocele because of recurrent urinary tract infections or persistent reflux (50). At times, preliminary cutaneous diversion of the involved ureter with delayed reconstruction is warranted, to allow maximal recovery of function before deciding on the need for nephrectomy vs. reconstruction (47).
Ureteropelvic Junction Obstruction
Obstruction at the ureteropelvic junction probably is the most common cause of a palpable abdominal mass in the newborn, and is the most common cause of antenatal hydronephrosis. This lesion usually is the result of narrowing of the ureter at the junction of the renal pelvis with the ureter. Because the renal pelvis is compliant, there can be a great deal of renal preservation despite massive dilation of the kidney behind the obstruction (Fig. 43-11) (54).
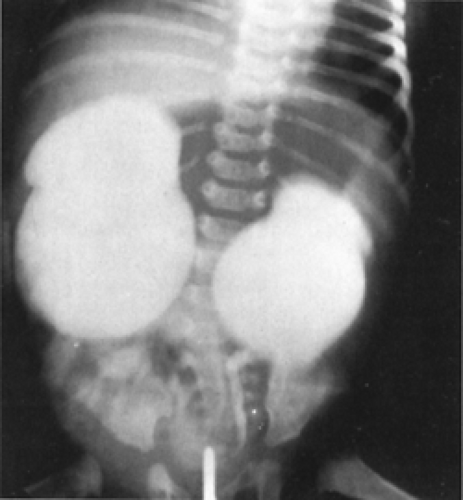 Figure 43-11 Severe bilateral hydronephrosis secondary to ureteropelvic junction obstructions. |
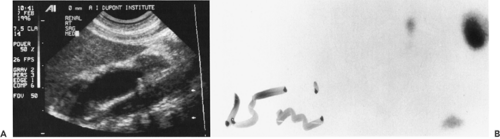 Figure 43-12 A: An abdominal ultrasonogram in a newborn infant demonstrates a single, large echo-free region consistent with hydronephrosis. B: A delayed renal scan in a newborn infant with right hydronephrosis secondary to ureteropelvic junction obstruction. |
The diagnosis of ureteropelvic junction obstruction can be made sonographically because there is a sonolucent central mass within the renal area surrounded by thin renal parenchyma (Fig. 43.12A). A grading system has been suggested by the Society for Fetal Urology to better standardize terminology and interpretation of renal ultrasound findings (55). The system is based on the severity of renal pelvis and calyceal dilatation and the renal parenchymal thickness (Fig. 43-13). Vesicoureteral
reflux must be excluded from the differential diagnosis using voiding cystography. The function of the obstructed kidney can be determined by radionuclide scanning (Fig. 43-12B). One of the advantages of radionuclide scintigraphy is that the physiologic significance of dilatation can be determined by administering furosemide (56). If the dilatation is of significance, there will be retention of the radionuclide behind the obstruction, whereas if there is no physiologic significance to the hydronephrosis, the administered diuretic will cause the radionuclide to wash out rapidly from the dilated system. To obtain valid results, the patient must be adequately hydrated and have adequate bladder drainage to prevent false interpretation of obstruction on the scan as a result of dehydration or bladder distention (57). It is becoming apparent, in some instances, that presumed significant hydronephrosis in the newborn is physiologically insigni-ficant and may with time, stabilize or improve and require no treatment. Those instances that are physiologically significant, may require repair, generally dismembered pyeloplasty, resulting in improved drainage and renal function in some instances (54).
reflux must be excluded from the differential diagnosis using voiding cystography. The function of the obstructed kidney can be determined by radionuclide scanning (Fig. 43-12B). One of the advantages of radionuclide scintigraphy is that the physiologic significance of dilatation can be determined by administering furosemide (56). If the dilatation is of significance, there will be retention of the radionuclide behind the obstruction, whereas if there is no physiologic significance to the hydronephrosis, the administered diuretic will cause the radionuclide to wash out rapidly from the dilated system. To obtain valid results, the patient must be adequately hydrated and have adequate bladder drainage to prevent false interpretation of obstruction on the scan as a result of dehydration or bladder distention (57). It is becoming apparent, in some instances, that presumed significant hydronephrosis in the newborn is physiologically insigni-ficant and may with time, stabilize or improve and require no treatment. Those instances that are physiologically significant, may require repair, generally dismembered pyeloplasty, resulting in improved drainage and renal function in some instances (54).
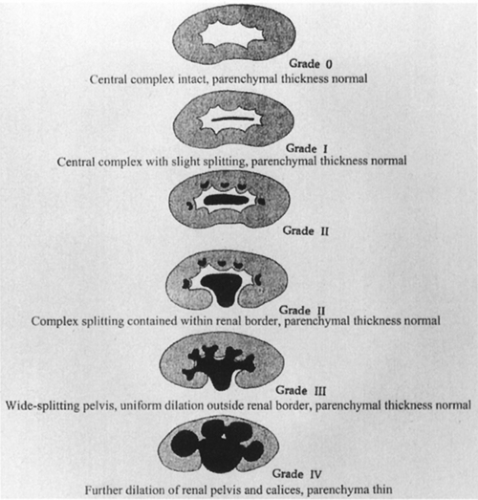 Figure 43-13 Society for Fetal Urology grading system for ultrasonographically detected hydronephrosis. From Baskin LS. Prenatal hydronephrosis. In: Baskin LS, Kogan BA, Duckett JW, eds. Handbook of pediatric urology. Philadelphia: Lippincott-Raven, 1997:11, with permission. Modified by Curt Powell, M.D. |
Ureterovesical Obstruction
Obstruction at the ureterovesical junction is not nearly so common as obstruction at the ureteropelvic junction, but it is far from rare (36). Lower ureteral obstruction may present as marked hydroureteronephrosis (i.e., a mass), but may sometimes present as urinary infection (Fig. 43-14). As with ureteropelvic obstruction, it has become apparent that not all ureterovesical obstructions are physiologically significant and some require no treatment. Radionuclide scanning with diuretics is helpful in making the diagnosis of a physiologic obstruction (56). At times, however, an antegrade pyelogram with a pressure perfusion study is necessary to determine the significance or lack of significance of an apparent narrowing at the ureterovesical junction (58). These lesions, if identified as obstructive, are treated by excision of the obstructing segment, tailoring or tapering of the dilated ureter, and reimplantation of the ureter into the bladder (59).
Vesicoureteral Reflux
Vesicoureteral reflux is the most common abnormality of the urinary system seen in children; it may occur in 1 of 100 births (12). The actual incidence is unknown, but it is at least as common as cryptorchidism or hypospadias. Genome mapping, performed on families with primary vesicoureteral reflux who also have associated reflux nephropathy, has found an association with a locus on chromosome 1 which is also seen in some families with vesicoureteral reflux (60). Vesicoureteral reflux is known to
be a familial problem. When one child in a family is identified as having reflux, as many as 30% to 50% of the siblings of that child may have vesicoureteral reflux (61). For that reason, it is thought that all younger siblings of any proband identified to have reflux should be screened (62). It also has been found that transmission of reflux from a previously refluxing parent to his or her children is approximately 66% (61). The normal ureterovesical junction is a relatively efficient mechanism that allows egress of urine into the lumen of the bladder, but, because of its oblique course through the ureteral wall, prevents the bladder urine from reentering the ureter (Fig. 43-15) (63). It is obvious that there is maturation of the ureterovesical junction with both time and growth, because infants have a much higher incidence of vesicoureteral reflux than do older children (64).
be a familial problem. When one child in a family is identified as having reflux, as many as 30% to 50% of the siblings of that child may have vesicoureteral reflux (61). For that reason, it is thought that all younger siblings of any proband identified to have reflux should be screened (62). It also has been found that transmission of reflux from a previously refluxing parent to his or her children is approximately 66% (61). The normal ureterovesical junction is a relatively efficient mechanism that allows egress of urine into the lumen of the bladder, but, because of its oblique course through the ureteral wall, prevents the bladder urine from reentering the ureter (Fig. 43-15) (63). It is obvious that there is maturation of the ureterovesical junction with both time and growth, because infants have a much higher incidence of vesicoureteral reflux than do older children (64).
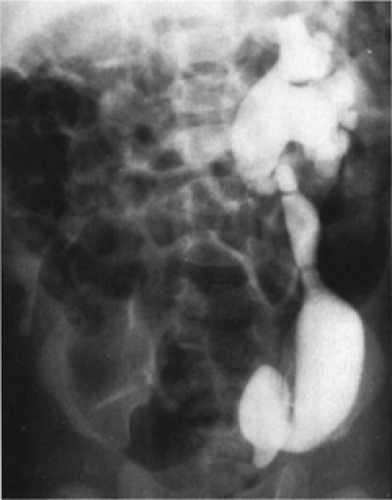 Figure 43-14 Left ureterovesical obstruction in a 3-month-old boy who presented with unresponsive diarrhea. Urine culture was positive, and radiographic evaluation demonstrated significant pathology, as is evident in this illustration. The diarrhea resolved with treatment of the urinary infection. The ureterovesical obstruction was treated surgically. From Kelalis PP, King LR, Belman AB, eds. Clinical pediatric urology, vol. 1. Philadelphia: WB Saunders, 1976:276, with permission. |
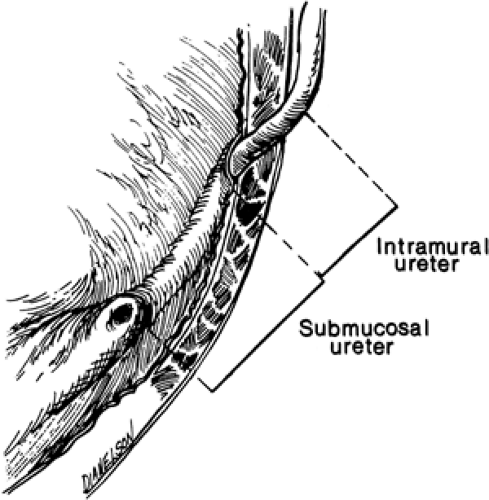 Figure 43-15 Normal ureterovesical junction. From Harrison JH, et al, eds. Campbell’s urology, 4th ed. Philadelphia: WB Saunders, 1979:1597, with permission. |
Reflux is graded on an international grading scale of 1 to 5 (65). The major significance of this grading system is that the higher the grade of reflux, the more likely it is that reflux will persist despite somatic growth, and the more likely that there is associated or eventual reflux nephropathy. Conversely, the lower the grade of reflux, the more likely there will be spontaneous resolution of the vesicoureteral reflux without reflux nephropathy (66).
Although the presence of hydronephrosis suggests an abnormality in the urinary tract, many patients with significant vesicoureteral reflux will have a normal ultrasound study. The radiologic confirmation of vesicoureteral reflux is accomplished by voiding cystourethrography. Generally, this should not be performed while the child is actively infected; if an infection has been the presenting sign, voiding cystography may be performed once the urine is sterile and the patient afebrile and under treatment. Because renal scarring is produced easily in the neonate, it is especially important to establish the presence or absence of vesicoureteral reflux before discontinuing antibiotics in patients who have presented with urinary infection.
Once reflux is demonstrated, especially in the infant, the patient should be maintained on low-dose antibacterial therapy until resolution of the reflux, hopefully preventing urinary tract infections. The choices of agents are limited in the newborn, but amoxicillin is a reasonable alternative until hepatobiliary maturation is sufficient to allow the use of sulfa or nitrofurantoin. A reasonable initial daily suppression dose would be 10 to 25 mg/kg of amoxicillin. With growing concerns of bacterial resistance to amoxicillin, low dose sulfamethoxasole-trimethoprim or nitrofurantoin may be used if carefully monitored. Breakthrough infection, while the patient is on antibacterial suppression or if there is poor parental compliance, suggests the need for surgical repair, either with open surgical techniques or endoscopic techniques.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


