Equinovarus – foot twisted inwards
Equinus – extended foot
Risk factors and clues to the diagnosis of skeletal anomalies
Family history
Although the majority of skeletal abnormalities are unexpected findings detected by ultrasound, some arise because of a relevant family history and others because of maternal drug use or maternal disease (Table 11.2). Whilst diagnosis can be more straightforward in families where there is an affected child or when one parent is affected with a dominantly-inherited condition, parents do need to be aware that some conditions, e.g., achondroplasia[3], may present relatively late in pregnancy, and others may be more variable, e.g., hypochondroplasia, and thus not necessarily amenable to sonographic diagnosis. For these reasons, molecular genetic diagnosis may be the best option for definitive diagnosis, but in most cases this will necessitate a detailed genetic work-up prior to pregnancy in order to identify relevant mutations. Molecular genetic diagnosis requires fetal tissue or DNA for analysis, which in the past has required an invasive test (chorionic villus sampling or amniocentesis). However, this is increasingly possible using noninvasive prenatal diagnosis (NIPD) and analysis of cell-free fetal DNA (cffDNA) in maternal plasma[4, 5]. In view of the rapid advances in molecular genetics, genetic advice should be sought before pregnancy in families of known high risk to be sure of the optimum method of diagnosis.
CNS, central nervous system; IUFGR, intrauterine fetal growth restriction.
Maternal drug ingestion or disease
There are a number of drugs that may be implicated in the etiology of fetal skeletal anomalies (Table 11.2). Maternal conditions, such as insulin-dependent diabetes, myasthenia gravis and myotonic dystrophy, can cause a variety of skeletal problems (Table 11.2), with other conditions, such as systemic lupus erythematosis and hypothyroidism also causing skeletal changes, but less commonly. In maternal myasthenia gravis, even when the mother is asymptomatic, transmission of acetycholine receptor antibodies to the fetus can result in generalized arthrogryposis and neonatal or infant death. In mothers with symptomatic myotonic dystrophy (an autosomal dominant condition), there is an up to 50% chance of the baby having congenital myotonic dystrophy (Table 11.2), which carries a high neonatal mortality (around 20%), with most survivors having significant developmental delay and reduced life expectancy.
Fetal limb development and the timing of diagnosis
An understanding of the timing of the development of the fetal skeleton is essential for accurate sonographic diagnosis, particularly as there is an increasing tendency towards detailed anomaly scanning in early pregnancy. In the human, the upper limbs develop a few days in advance of the lower limbs, with the arm buds appearing at about 5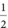 postmenstrual weeks. The clavicle begins to ossify at around 8 weeks’ gestation, followed by the mandible, vertebral bodies and neural arches around 9 weeks, the frontal bones at 10–11 weeks and the long bones from 8 weeks (Figure 11.1)[6]. Of note, ossification of the cervical and sacral spine is not complete until around 20 weeks’ gestation. Most skeletal structures can be identified by 12 weeks’ gestation, but transvaginal scanning is recommended for accurate diagnosis at this gestation. In addition to sonographic expertise, accurate identification of skeletal anomalies requires aids such as charts of normal skeletal size, including length of long bones, clavicles, mandible, scapular, chest size, orbital diameters, renal size, etc.[7–10]. Given knowledge of skeletal development, sonographic expertise and necessary aids, and with the technologic advances in ultrasound platforms, it is clear that scanning in the first and early second trimesters can be ideal for the detection of many serious skeletal dysplasias (Tables 11.3 and 11.4;[11]) as well as localized limb reduction defects (Table 11.5;[12]).
postmenstrual weeks. The clavicle begins to ossify at around 8 weeks’ gestation, followed by the mandible, vertebral bodies and neural arches around 9 weeks, the frontal bones at 10–11 weeks and the long bones from 8 weeks (Figure 11.1)[6]. Of note, ossification of the cervical and sacral spine is not complete until around 20 weeks’ gestation. Most skeletal structures can be identified by 12 weeks’ gestation, but transvaginal scanning is recommended for accurate diagnosis at this gestation. In addition to sonographic expertise, accurate identification of skeletal anomalies requires aids such as charts of normal skeletal size, including length of long bones, clavicles, mandible, scapular, chest size, orbital diameters, renal size, etc.[7–10]. Given knowledge of skeletal development, sonographic expertise and necessary aids, and with the technologic advances in ultrasound platforms, it is clear that scanning in the first and early second trimesters can be ideal for the detection of many serious skeletal dysplasias (Tables 11.3 and 11.4;[11]) as well as localized limb reduction defects (Table 11.5;[12]).

Chart demonstrating gestation at which ossification of the fetal skeleton commences as indicated from radiologic studies of fetuses after delivery (adapted from Calder and Offiah[6]).
| Examination | Outcome | Common diagnoses | Other investigationsa |
|---|---|---|---|
| Measure all long bones | All short | IUFGR | Maternal and fetal Dopplers. Review other biometry for small HC/AC. Serial scanning for growth velocity. Review maternal serum screening results for high hCG and MSAFP or low PAPP-A. Obstetric history for IUFGR, SB, PET, etc. Maternal medical history for autoimmune disease. Check for reduced AFI. |
| Constitutional short stature | F/H, examine parents, normal long bone growth velocity, normal Dopplers. | ||
| Aneuploidy | Detailed anomaly scan for markers of aneuploidy. Consider karyotyping. | ||
| Skeletal dysplasia | See below | ||
| Assess the gestation at onset of shorteningb | First trimester | Thanatophoric, OI IIA/C/B, achondrogenesis types 1 and 2, most SRPSs, diastrophic dysplasia, SEDC, hypophosphatasia, Ellis–van Creveld, Boomerang dysplasia | |
| Second trimester | OI IIB/III/IV, Jeune’s asphyxiating thoracic dystrophy, most chondrodysplasia punctatas | ||
| Third trimester | Achondroplasia | Exclude IUFGR, maternal blood for NIPD for FGFR3 mutations. | |
| Compare measurements of proximal and distal long bones to classify type of shortening | Rhizomelic – shortening most evident in proximal long bones (humeri and femora) | Diastrophic dysplasia, SEDC, Jeune’s asphyxiating thoracic dystrophy | |
| Mesomelic – shortening of the mid-section of a limb (radius/ulna, tibia/fibula) | Ellis–van Creveld, Achondroplasia, oral-facial-digital IV | ||
| Micromelic – all long bones are very short | Achondrogenesis type 1 and 2, Boomerang dysplasia | ||
| Evaluate the structure of bones | Fractured/bowed | OI type IIA/B/C/III/IV. Hypophosphatasia, campomelic dysplasia (legs only) | Parental alkaline phosphatase levels, urinary phosphoethanolamine levels. |
| Hypomineralized | OI type IIA/B/C/III Hypophosphatasia | ||
| Metaphyseal flaring | Kniest syndrome | ||
| Absent bones | Roberts syndrome | Chromosome analysis for centromeric puffing. | |
| Stippled epiphyses | Rhizomelic chondrodyplasia punctata, Conradi Hunermann, X-linked recessive chondrodyplasia punctate, warfarin embryopathy, maternal SLE/auto-immune disease | Maternal drug history, mutations in ARSE gene, metabolic investigations – very long chain fatty acids and sterol profile, maternal history of autoimmune disease. | |
| Examine the extremities | Polydactyly | Jeune’s asphyxiating thoracic dystrophy, Ellis–van Creveld syndrome, SRPSs | Family history, check for consanguinity. |
| Syndactyly | Apert syndrome | NIPD for FGFR2 mutations. | |
| Polysyndactyly | SRPSs, oral-facial-digital IV | ||
| Oligodactyly | De Lange, Roberts syndrome | ||
| Radial club hand | VATER/VACTERL | ||
| Short fingers/trident hand | Achondroplasia, acromesomelic dysplasia, thanatophoric dysplasia | Screen for mutations in FGFR3 gene. | |
| Talipes | Diastrophic dysplasia, campomelic dysplasia, Kniest, oral-facial-digital IV | ||
| Hitchhiker thumbs/toes | Diastrophic dysplasia | ||
| Examine the skull – size and shape | Severe hypomineralisation | OI IIA/C, achondrogenesis type 1, hypophosphatasia (severe neonatal type), Boomerang dysplasia | |
| Mild hypomineralisation | OI IIB (OI III), cleido-cranial dysostosis | Examine the parents for cleido-cranial dysostosis. | |
| Cloverleaf skull or craniosynostosis | Thanatophoric dysplasia II, occasionally in SRPSs, Apert, craniosynostosis syndromes | Maternal blood for NIPD to screen cfDNA for mutations in the FGFR3 and 2 genes. | |
| Relative macrocephaly | Thanatophoric dysplasia, achondroplasia, achondrogenesis type 1 | Maternal blood for NIPD to screen cfDNA for mutations in the FGFR3. | |
| Progressive microcephaly | Rhizomelic chondrodysplasia punctate | ||
| Examine the clavicles and scapulae | Short/absent clavicles | Cleido-cranial dysostosis | Examine parents. |
| Small scapula | Campomelic dysplasia | NIPT for fetal sex if genitalia are ambiguous or female. | |
| Examine the face – profile and coronal views | Frontal bossing | Achondroplasia, thanatophoric dysplasia, acromesomelic dysplasia | Maternal blood for NIPD to screen cfDNA for mutations in the FGFR3 gene. |
| Micrognathia | SEDC, Stickler syndrome, campomelic dysplasia, diastrophic dysplasia, Kniest syndrome | Examine parents. Consider NIPD for fetal sex if genitalia are ambiguous or female. | |
| Cleft lip | SRPSs – Majewski syndrome, Verma–Naumoff and Beemer–Langer, Ellis–van Creveld, Roberts syndrome, oral-facial-digital IV | ||
| Absent/flat nasal bridge | Rhizomelic chondrodysplasia punctata, Binder phenotype, Warfarin embryopathy | Drug history, mutations in arylsulfatase E gene, metabolic investigations – very long chain fatty acids and sterol profile, maternal history of autoimmune disease. | |
| Mid-face hypoplasia | Rhizomelic chondrodysplasia punctata, Binder phenotype, Kniest | ||
| Small nose | Rhizomelic chondrodysplasia punctata, Binder phenotype, Kniest | ||
| Cataracts | Rhizomelic chondrodysplasia punctate | ||
| Examine the thorax – assess the size by measuring circumference by comparing with the abdomen in the axial plane or assessing lung:heart ratio. Examine the parasagittal view for the ‘champagne-cork’ appearance | Short | SEDC, Stickler, Kniest | Examine the parents. |
| Very small and narrow with champagne cork appearance | Thanatophoric dysplasia, SRPSs, dystrophy achondrogenesis, OI IIA/C | ||
| Long and narrow | Jeune’s asphyxiating thoracic dystrophy. Occasionally achondroplasia and Ellis–van Creveld | ||
| Examine the ribs in the axial plane for length and sagittal/coronal plane for beading/fractures/missing ribs | Short straight ribs | Thanatophoric dysplasia, SRPSs, Jeune’s asphyxiating thoracic dystrophy, achondrogenesis type 2, Ellis–van Creveld , achondrogenesis type 1 | |
| Fractures/beaded ribs | OI IIA/C/B | ||
| Absent/disorganized ribs | Jarcot–Levin, spondylocostal dysplasia, campomelic dysplasia (occasionally only 11 ribs). | ||
| Examine the spine for mineralization, disorganization, hemivertebrae | Hypomineralized vertebral bodies | Achondrogenesis type I | |
| Hemivertebrae | VATER/VACTERL Spondylocostal dysplasia | ||
| Disorganization (may be confused with extra calcification) | Jarcot–Levin, spondylocostal dysplasia, some chondrodysplasia punctatas, dyssegmental dysplasia, VATER/VACTERL | ||
| Examine the skin for edema | Increased nuchal translucency | Thanatophoric, OI, SRPSs, SEDC and several others | |
| Hydrops | SRPSs, achondrogenesis type 1, Boomerang dysplasia | ||
| Examine the fetal viscera | CNS anomaly | SRPSs (Majewski, Beemer–Langer), thanatophoric dysplasia type II, occasionally achondroplasia | |
| Cardiac anomaly | Campomelic dysplasia, Ellis van Creveld, SRPSs, VATER/VACTERL | ||
| Renal tract anomaly | Jeune’s asphyxiating thoracic dystrophy, SRPSs | ||
| Anterior abdominal wall defect | SRPS (Beemer–Langer) | ||
| Genital anomalies | Campomelic dysplasia, SRPSs | NIPD for fetal sex determination | |
| Amniotic fluid volume | Polyhydramnios | Achondroplasia, thanatophoric dysplasia, paternal uniparental disomy 14, VATER/VACTERL | |
| Oligohydramnios | SRPSs with renal anomalies. |
a Investigations recommended if other features are compatible with a diagnosis.
b There is variability for some dysplasias and the timings given here reflect the authors’ personal experience. In addition, where there is a family history scanning may be targeted at certain features that may otherwise be overlooked on a routine scan facilitating earlier diagnosis.
AC: abdominal circumference; AFI: amniotic fluid index; cfDNA: cell-free DNA; CNS: central nervous system; FGFR3: fibroblast growth factor 3; HC: head circumference; hCG: human chorionic gonadotropin; IUFGR: intrauterine fetal growth restriction; MSAFP: maternal serum alpha-fetoprotein; SRPS: short-ribbed polydactyly syndrome; NIPT: noninvasive prenatal testing; OI: osteogenesis imperfecta; PAPP-A: pregnancy-associated plasma protein A: PET: pre-eclamptic toxemia; SB: stillbirth; SEDC: spondylo-epiphyseal dysplasia congenica; SLE: systemic lupus erythema; VATER/VACTERL: vertebral anal atresia tracheoesophageal fistula radial anomaly/vertebral anal atresia cardiac trachea-esophageal fistula radial limb anomaly.
Adapted from reference 12. AD, autosomal dominant; AR, autosomal recessive FL, femur length; HC, head circumference; NT, nuchal translucency; SRPS, short-ribbed polydactyly syndrome.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


