Group
Diagnostic criteria
1–8
Based on a common underlying gene or pathway (e.g., achondroplasia belongs to group 1 or the FGFR3 chondrodysplasia group)
9–17
Based on the localization of radiographic changes to specific bone structures (vertebrae, epiphyses, metaphyses, diaphyses, or a combination thereof) or of the involved segment (rhizo, meso, or acro)
18–20
Defined by macroscopic criteria in combination with clinical features (bent bones, slender bones, presence of multiple dislocations)
21–25, 28
Takes into account features of mineralization (increased or reduced bone density, impaired mineralization, stippling, osteolysis) (e.g., osteogenesis imperfecta and other disorders of decreased bone density belong to group 25)
26
Identification of several novel molecular mechanisms leading to hypophosphatemic rickets
27
Lysosomal disorders with skeletal involvement
29
Disorders with so-called abnormal development of skeletal components (exostoses, enchondromas, and ectopic calcification)
30
Overgrowth syndromes with significant skeletal involvement
31
Genetic inflammatory/rheumatoid-like osteoarthropathies
32–40
Dysostoses, anatomical criteria with additional criteria reflecting principles of embryonic development such as limb reduction or hypoplasia (proximal-distal growth) versus terminal differentiation and patterning of the digits or joint formation
Pathogenetics
Recent advances in the understanding of the various causes of SD have been due in large part to the study of the underlying molecular mechanisms and gene mutations (Superti-Furga et al. 2001). SD can be broadly grouped on the basis of the function of the protein product of the causative gene. This can be clinically relevant as many of the disorders caused by genes whose protein products have similar functions also share similar clinical characteristics. Five broad groups can be categorized based on the defective molecular component and proteins/genes involved as shown in Table 2 below.
Table 2
Pathogenetics of SD: 5 broad groups based on the defective molecular component and proteins/genes involved
Gene or protein involved | Clinical phenotype |
|---|---|
Group 1: defect in structural cartilage proteins | |
Collagen 2 (COL2A1) | Achondrogenesis 2, spondyloepiphyseal dysplasia, Kniest dysplasia, Stickler syndrome |
Group 2: defect in cartilage metabolic pathways | |
Diastrophic dysplasia sulfate transporter | Diastrophic dysplasia, achondrogenesis IB, atelosteogenesis type 2, recessive multiple epiphyseal dysplasia |
Group 3: defect in local regulators of cartilage growth | |
Fibroblast growth factor receptor 3 (FGFR3) | Achondroplasia, thanatophoric dysplasia, hypochondroplasia |
Group 4: defect in transcription factors | |
Short-stature homeobox gene (SHOX) | Dyschondrosteosis, Langer-type mesomelic dysplasia |
Group 5: defect in tumor-suppressor genes | |
Exostosin 1 and 2 | Multiple hereditary exostoses |
Many of the genes mutated in SD affect important functions of the growth plate (physis). A good understanding of growth plate physiology will be helpful in working out the possible effects of gene mutations in various zones of the physis. For example, in the resting zone, SOX9 gene mutation causes camptomelic dysplasia; in the proliferative zone, FGFR3 gene mutation causes achondroplasia, hypochondroplasia, and thanatophoric dysplasia; in the hypertrophic zone, PTHR1 gene mutation causes metaphyseal dysplasia; and in the terminal differentiation zone, RUNX2 gene mutation causes cleidocranial dysplasia (Dietz and Mathews 1996; Zelzer and Olsen 2003).
Diagnosis
The diagnosis of SD usually requires a multidisciplinary approach with the active participation of a pediatric geneticist, a radiologist, and a molecular pathologist with special interest in SD as well as a pediatric orthopedic surgeon.
Prenatal Diagnosis
Long-bone biometry has been used extensively in the prediction of the fetal gestational age. Nomograms generally use the long bone as the independent variable and the estimated fetal age as the dependent variable. However, in order to assess the normality of bone dimensions, the gestational age is used as the independent variable and the long bone as the dependent variable. Patients at risk for SD should seek prenatal care early to assess all clinical estimators of gestational age. In the prenatal diagnosis of infants with SD, there is a discrepancy between fetal size and gestational age. Affected fetuses have been shown to have dramatic deviations from the 5th and 95th confidence limits (Romero et al. 1998). The graphs below show the biometry of the femur and humerus during gestation (Figs. 1 and 2).
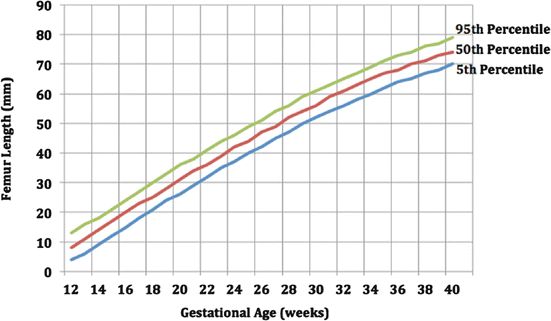
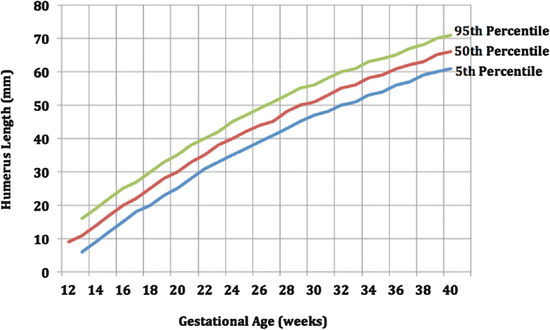
Fig. 1
Biometry of femur during gestation
Fig. 2
Biometry of humerus during gestation
Postnatal Diagnosis
History
When presented with a child with disproportionate short stature, a focused history can give important clues as to the differential diagnosis. A complete prenatal history should be obtained, including fetal biometry findings during the prenatal ultrasound.
As a part of the patient’s perinatal history, it is important to ascertain the patient’s birth length, as some patients with skeletal dysplasia may present with short stature at birth (e.g., achondroplasia) while others may have a normal birth length with subsequent failure of linear growth (e.g., pseudoachondroplasia).
Family Evaluation
It is important to take a detailed family history to determine if there is another family member with SD in order to assess the mode of inheritance, if any. SD is often transmitted by Mendelian inheritance. A pedigree that includes first-degree relatives is usually sufficient for screening, but occasionally, a more extensive family history is needed. Assessment of parental heights also plays a role, as the child simply might have familial short stature (Unger 2002).
Physical Examination
Growth parameters should be measured, which include the patient’s height, weight, and head circumference. For example, in patients with achondroplasia, the head circumference is greater than normal while the height is reduced dramatically. It is also important to note the height for age percentile of the patient. In general, if adult height is under 150 cm or 5 ft, consideration of SD is appropriate.
It is also necessary to determine the patient’s proportions. The upper segment to lower segment ratio and the arm span to height ratio are used to document whether the spine or the limb is more severely shortened. Limb shortening is further classified depending on which segment is most affected: rhizomelia (short proximal segments; i.e., short humerus or femur, as in achondroplasia), mesomelia (short middle segments; i.e., short forearm or tibia, as in dyschondrosteosis), or acromelia (short hands or feet, as in Grebe type of chondrodystrophy) (Fig. 3).
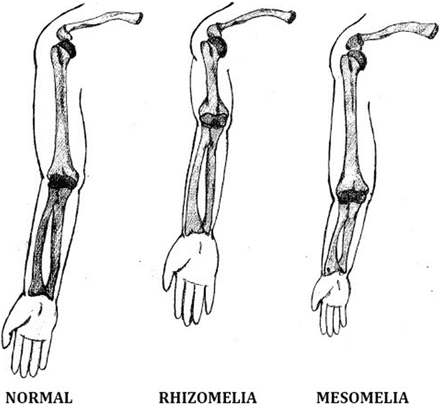
Fig. 3
Types of disproportionate limb shortening
When the patient is short but the body proportions are normal, the diagnosis is usually related to constitutional short stature, which may be due to endocrine disorders, malnutrition, prenatal dwarfism, or one of the many dysmorphic syndromes.
Patients with SD often have dysmorphism or minor morphologic variations of the bones and soft tissues. Dysmorphic features associated with short stature suggest an underlying SD (Beals and Horton 1995).
The presence of deformities (e.g., spinal deformities such as lordosis, kyphosis, or scoliosis; varus or valgus deformity of the extremities) in patients with short stature is also highly suggestive of a diagnosis of an SD.
Imaging Studies
Good-quality skeletal radiographs should be obtained. It is necessary to request for a limited skeletal survey because normal findings in a specific region can aid in making a differential diagnosis. An SD can almost always be diagnosed on the basis of five radiographs: lateral skull, anteroposterior (AP) view of the pelvis, lateral lumbar spine, AP view of the hand and wrist, and AP view of the knee. In general, the dysplasias are classified according to which part of the skeleton is involved. The pattern of involvement may include any or all of the following: spondyloepiphyseal, metaphyseal, and diaphyseal dysplasia. Aside from the pattern of involvement, the region affected can also be used to narrow the differential diagnosis (Unger 2002).
Other Tests
Many of the SD can be diagnosed from clinical and radiographic features. However, a blood chemistry profile can be obtained to exclude treatable metabolic disorders such as the mucopolysaccharidoses and rickets. A bone biopsy is rarely indicated but may sometimes be useful to confirm the diagnosis of dysplasia. In some instances, more detailed genetic testing may be required for purposes of confirmation of the diagnosis, documentation, and counseling.
Differential Diagnosis
In considering the differential diagnosis, a few important conditions should come to mind. These are hypothyroidism, nutritional rickets, failure to thrive, and non-accidental injuries.
General Approach to Treatment
A multidisciplinary approach is usually required to manage SD as the children often have multiple problems that require different areas of expertise. Genetic counseling is an important aspect of management and a pediatric geneticist would be the ideal person to conduct this appraisal. In centers where antenatal diagnostic technology is available, preimplantation genetic diagnosis could help the parents to have a normal child. In addition to genetic counseling, the parents and child will benefit from counseling with respect to potential medical and social problems that the child could encounter during growth and development.
More specific to the type of SD, the child will require management for spine and limb deformities, possible fractures (e.g., in osteogenesis imperfecta), limb length discrepancy, and in some situations limb lengthening for short stature. Some of these will be covered in greater detail in the sections below on Achondroplasia and Osteogenesis Imperfecta.
Commonly Seen Conditions
Achondroplasia
Achondroplasia is the most common SD, with an incidence of 1 in 30,000 live births per year (Shirley and Ain 2009). It is inherited as a fully penetrant autosomal dominant trait. However, more than 80 % of the cases are sporadic, wherein affected individuals are the offspring of parents of normal stature. Higher paternal age at the time of conception of patients with achondroplasia suggests that de novo mutations of paternal origin are involved (Vajo et al. 2000).
Pathogenetics
Achondroplasia is caused by mutations of the gene encoding fibroblast growth factor receptor 3 (FGFR3) on the distal short arm of chromosome 4. Most individuals with the clinical features of achondroplasia have the same mutations that substitute an arginine for a glycine residue in the transmembrane domain of the receptor in the physis. FGFR3 is a negative regulator of chondrocyte proliferation and differentiation in the growth plate. Mutations involving the FGFR3 result in activation of the receptor and thus viewed as a gain-of-function mutation (Vajo et al. 2000; Horton 2006; Laederich and Horton 2010).
The clinical result of this mutation is underdevelopment and shortening of the long bones formed by endochondral ossification. It has been suggested that FGFR3 inhibits both the proliferation and terminal differentiation of growth plate chondrocytes and synthesis of extracellular matrix by these cells. It also proposed that FGFR3 induces premature terminal differentiation, thereby reducing the number of cells that contribute to template synthesis (Laederich and Horton 2010).
Prognosis
Affected individuals have normal cognitive development, although motor development may be delayed. Patients with achondroplasia are healthy compared to patients with other skeletal dysplasias, but mortality rates in all age groups are higher than those in the general population because of sudden death in young infants, central nervous system and respiratory problems in older children, and cardiovascular problems in young adults.
Diagnosis
The clinical manifestations of achondroplasia vary at different stages of the affected individual’s life.
At birth, short stature is evident, with the trunk length in the lower range of normal and the extremities shortened in a rhizomelic pattern. Frontal bossing and midface hypoplasia are also noted. Delayed motor development or apnea in infants may be the result of foramen magnum stenosis leading to cervical myelopathy. Hydrocephalus may occur during the newborn and infantile period.
As the infant grows and begins to sit, thoracolumbar kyphosis may be noted. This is often due to the infant slumping forward because of trunk hypotonia combined with a relatively oversized head, a flat chest, and a protuberant abdomen. This decreases with age (most tend to resolve at 12–18 months of age) as trunk strength improves and the child begins to walk.
As the child continues to grow, genu varum and lumbosacral hyperlordosis become evident. The presence of genu varum is a clinical hallmark of achondroplasia. Its cause remains controversial, with several hypotheses being proposed such as lateral collateral ligament laxity and fibular overgrowth. Lumbosacral hyperlordosis is the result of excessive anterior pelvic tilt while standing, producing a prominent abdomen and buttocks with hip flexion contractures. In adulthood, lumbar spinal stenosis may develop.
The rhizomelic shortening of the upper extremity of affected individuals can create disability because of the difficulty in reaching the top of the head and the perineum for hygiene care. This can also be exacerbated by elbow flexion contractures and radial head subluxation.
The average final height of adult individuals with achondroplasia is 132 cm for males (range: 118–145 cm) and 125 cm for females (range: 112–136 cm). This corresponds to a height that is 6–7 standard deviations below the average for unaffected individuals (Shirley and Ain 2009).
Management
Management of achondroplasia is directed toward its different clinical manifestations. Indications for surgery for the spinal conditions associated with achondroplasia are not clearly defined, although neurologic compromise generally warrants appropriate surgical intervention. The decision for limb lengthening is likewise difficult and controversial, as the lengthening process is long, arduous, and complicated. In addition, the functional improvement after elective limb lengthening has not been well established.
Osteogenesis Imperfecta
Osteogenesis imperfecta (OI) is a heritable heterogeneous disorder of bone formation that may affect more than 1 in 10,000 individuals. It is distinguished by four features that present in the following causal relationship: genetic disorder, collagen defect, bone fragility, and frequent fracture. Patients with OI are characterized by having fragile bones. Many also have dentinogenesis imperfecta (teeth appear brownish or bluish, soft, translucent, prone to cavities, and crack easily), blue sclerae (the effect of light reflected from the underlying choroid and its blood vessels through the thin sclerae), and scoliosis (Roughley et al. 2003). Minimal trauma can result in fractures and bony deformities.
Pathogenetics
OI is a group of disorders caused by inherited or spontaneous genetic mutations in the COL1A1 or COL1A2 genes, which are responsible for encoding the alpha-1 and alpha-2 chains of type 1 collagen, respectively. Quantitatively or qualitatively deficient fibrils are produced by mutations in these genes, resulting in bone tissue with altered mechanical properties. Bone tissue anomalies are the most visible manifestation of OI. Extraskeletal tissues and organs affected by type 1 collagen defects include the sclerae, dentine, ear ossicles, skin, vessels, capillaries, and heart valves.
Classification
In 1979, Sillence classified the condition into four types based on clinical, genetic, and radiographic findings. Since then, three more types have been added. Types I to IV are commonly associated with mutations in the genes for type 1 collagen, whereas no type 1 collagen gene mutation has been detected in types V to VII (Kocher and Shapiro 1998; Burnei et al. 2008).
Type I
Type I is the most common form of OI, estimated at 3–5 per 100,000 births. It is mild in severity and is inherited in an autosomal dominant pattern. Patients have blue sclerae and bone fragility and are deaf or have a family history of presenile deafness. Infants are of normal weight and length at birth and do not have multiple fractures. This type is subdivided into type A and type B according to the absence or presence of dentinogenesis imperfecta, respectively.
Type II
This is the lethal form of OI, and the pattern of inheritance is also autosomal dominant. However, the lack of affected siblings in different series suggests a new mutation of a dominant gene or a nongenetic etiology. Its incidence is 1 per 40,000–60,000 births. Infants are either stillborn or die during the neonatal period and are frequently small for gestational age. Blue sclerae are present. In utero, multiple fractures occur and the long bones are broad and shortened.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


