Chapter 586 Seizures in Childhood
A seizure is a transient occurrence of signs and/or symptoms resulting from abnormal excessive or synchronous neuronal activity in the brain. The International Classification of Epileptic Seizures divides epileptic seizures into 2 large categories: In focal (partial) seizures, the first clinical and electroencephalographic (EEG) changes suggest initial activation of a system of neurons limited to part of one cerebral hemisphere; in generalized seizures, the first clinical and EEG changes indicate synchronous involvement of all of both hemispheres (Table 586-1). Approximately 30% of patients who have a first afebrile seizure have later epilepsy; the risk is about 20% if neurologic exam, EEG, and neuroimaging are normal. Febrile seizures are a special category. Acute symptomatic seizures occur secondary to an acute problem affecting brain excitability such as electrolyte imbalance or meningitis. Most children with these types of seizures do well, but sometimes such seizures signify major structural, inflammatory, or metabolic disorders of the brain, such as meningitis, encephalitis, acute stroke, or brain tumor; the prognosis depends on the underlying disorder, including its reversibility or treatability and the likelihood of developing epilepsy from it. Unprovoked seizure is not an acute symptomatic seizure. Remote symptomatic seizure is thought to be secondary to a distant brain injury such as an old stroke.
Table 586-1 TYPES OF EPILEPTIC SEIZURES
SELF-LIMITED SEIZURE TYPES
Focal Seizures
Generalized Seizures
CONTINUOUS SEIZURE TYPES
Generalized Status Epilepticus
Focal Status Epilepticus
PRECIPITATING STIMULI FOR REFLEX SEIZURES
From International League Against Epilepsy: Epileptic seizure types and precipitating stimuli for reflex seizures (website), May 13, 2009. http://www.ilae-epilepsy.org/Visitors/Centre/ctf/seizure_types.cfm. Accessed October 8, 2010.
An epileptic syndrome is a disorder that manifests one or more specific seizure types and has a specific age of onset and a specific prognosis. Several types of epileptic syndromes can be distinguished (Tables 586-2 to 586-4). This classification has to be distinguished from the classification of epileptic seizures that refers to single events rather than to clinical syndromes. In general, seizure type is the primary determinant of the type of medications the patient is likely to respond to, and the epilepsy syndrome determines the type of prognosis one could expect. An epileptic encephalopathy is an epilepsy syndrome in which the severe EEG abnormality is thought to result in cognitive and other impairments in the patient. Idiopathic epilepsy is an epilepsy syndrome that is genetic or presumed genetic and in which there is no underlying disorder affecting development or other neurologic function (e.g., petit mal epilepsy). Symptomatic epilepsy is an epilepsy syndrome caused by an underlying brain disorder (e.g., epilepsy secondary to tuberous sclerosis). A cryptogenic epilepsy (also termed presumed symptomatic epilepsy) is an epilepsy syndrome in which there is a presumed underlying brain disorder causing the epilepsy and affecting neurologic function, but the underlying disorder is not known.
Table 586-2 CLASSIFICATION FOR EPILEPSY SYNDROMES WITH AN INDICATION OF AGE OF ONSET, DURATION OF ACTIVE EPILEPSY, PROGNOSIS, AND THERAPEUTIC OPTIONS
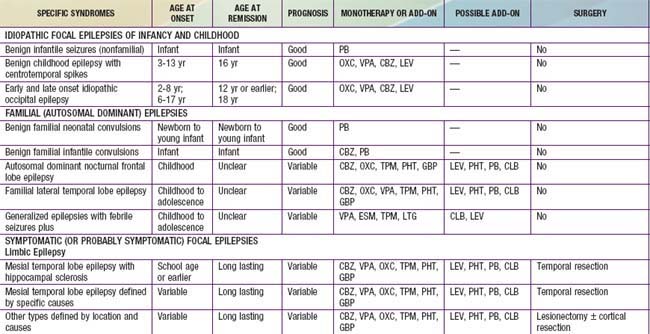
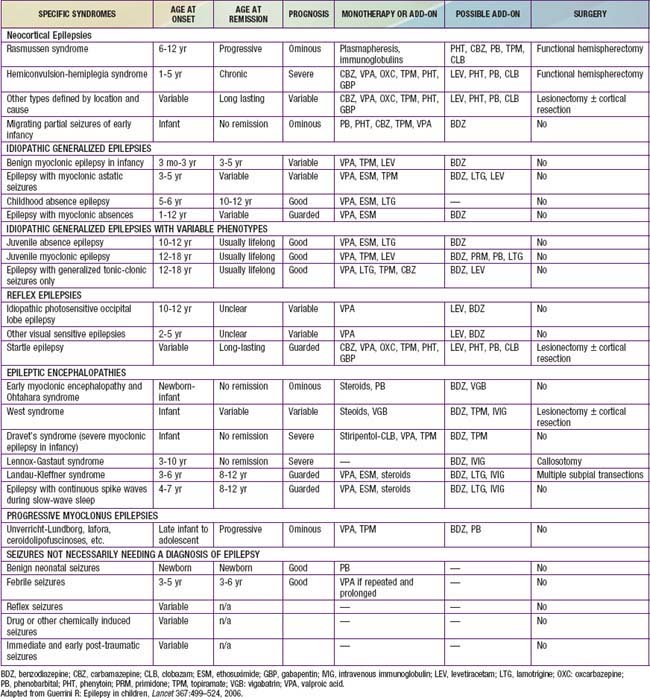
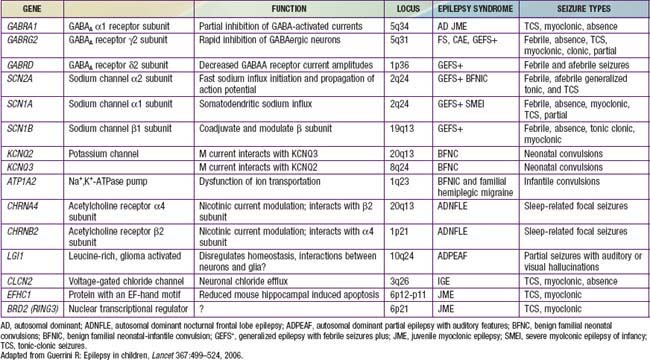
Table 586-4 CHILDHOOD EPILEPTIC SYNDROMES WITH GENERALLY GOOD PROGNOSIS
| SYNDROME | COMMENT |
|---|---|
| Benign neonatal familial convulsions | Dominant, may be severe and resistant during a few days Febrile or afebrile seizures (benign) occur later in a minority |
| Infantile familial convulsions | Dominant, seizures often in clusters (overlap with benign partial complex epilepsy of infancy) |
| Febrile convulsions plus syndromes (see Table 586-2) | In some families, febrile and afebrile convulsions occur in different members, GEFS+ The old dichotomy between febrile convulsions or epilepsy does not always hold |
| Benign myoclonic epilepsy of infancy | Often seizures during sleep, one rare variety with reflex myoclonic seizures (touch, noise) |
| Partial idiopathic epilepsy with rolandic spikes | Seizures with falling asleep or on awakening; focal sharp waves with centrotemporal location on EEG; genetic |
| Idiopathic occipital partial epilepsy | Early childhood form with seizures during sleep and ictal vomiting; can occur as status epilepticus Later forms with migrainous symptoms; not always benign |
| Petit mal absence epilepsy | Cases with absences only, some have generalized seizures. 60-80% full remission In most cases, absences disappear on therapy but there are resistant cases (unpredictable) |
| Juvenile myoclonic epilepsy | Adolescence onset, with early morning myoclonic seizures and generalized seizures during sleep; often history of absences in childhood |
EEG, electroencephalogram; GEFS+, generalized epilepsy with febrile seizures plus.
From Deonna T: Management of epilepsy, Arch Dis Child 90:5–9, 2005.
586.1 Febrile Seizures
Between 2% and 5% of neurologically healthy infants and children experience at least 1, usually simple, febrile seizure. Simple febrile seizures do not have an increased risk of mortality even though they are concerning to the parents. Complex febrile seizures may have an approximately 2-fold long-term increase in mortality, as compared to the general population over the subsequent 2 yr, probably secondary to coexisting pathology. There are no long-term adverse effects of having ≥1 simple febrile seizures. Specifically, recurrent simple febrile seizures do not damage the brain. Compared with age-matched controls, patients with febrile seizures do not have any increase in incidence of abnormalities of behavior, scholastic performance, neurocognitive function, or attention. Children who develop later epilepsy might experience such difficulties. Febrile seizures recur in approximately 30% of those experiencing a first episode, in 50% after 2 or more episodes, and in 50% of infants <1 yr old at febrile seizure onset. Several factors affect recurrence risk (Table 586-5). Although about 15% of children with epilepsy have had febrile seizures, only 2-7% of children who experience febrile seizures proceed to develop epilepsy later in life. There are several predictors of epilepsy after febrile seizures (Table 586-6).
Table 586-5 RISK FACTORS FOR RECURRENCE OF FEBRILE SEIZURES
MAJOR
MINOR
Having no risk factors carries a recurrence risk of about 12%; 1 risk factor, 25-50%; 2 risk factors, 50-59%; 3 or more, 73-100%.
Modified from Mikati MA, Rahi A: Febrile seizures: from molecular biology to clinical practice, Neurosciences 10:14–22, 2004.
Table 586-6 RISK FACTORS FOR OCCURRENCE OF SUBSEQUENT EPILEPSY
| RISK FACTOR | RISK FOR SUBSEQUENT EPILEPSY |
|---|---|
| Simple febrile seizure | 1% |
| Neurodevelopmental abnormalities | 33% |
| Focal complex febrile seizure | 29% |
| Family history of epilepsy | 18% |
| Fever <1 hr before febrile seizure | 11% |
| Complex febrile seizure, any type | 6% |
| Recurrent febrile seizures | 4% |
Modified from Mikati MA, Rahi A: Febrile seizures: from molecular biology to clinical practice, Neurosciences 10:14–22, 2004.
Work-Up
The general approach the patient with febrile seizures is delineated in Figure 586-1. Each child who presents with a febrile seizure requires a detailed history and a thorough general and neurologic examination. These are the cornerstones of the evaluation. Febrile seizures often occur in the context of otitis media, roseola and human herpesvirus 6 (HHV6) infection, shigella, or similar infections, making the evaluation more demanding. Several investigations need to be considered.
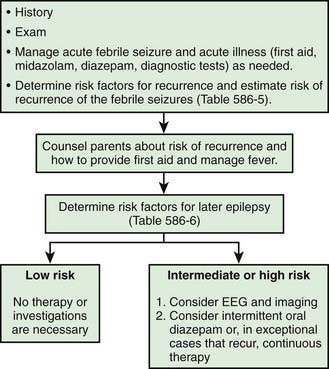
Figure 586-1 Management of febrile seizures.
(Modified from Mikati MA, Rahi A: Febrile seizures: from molecular biology to clinical practice, Neurosciences 10:14–22, 2004.)
Treatment
In general, antiepileptic therapy, continuous or intermittent, is not recommended for children with one or more simple febrile seizures. Parents should be counseled about the relative risks of recurrence of febrile seizures and recurrence of epilepsy, educated on how to handle a seizure acutely, and given emotional support. If the seizure lasts for >5 min, then acute treatment with diazepam, lorazepam, or midazolam is needed (see Chapter 586.8 for acute management of seizures and status epilepticus). Rectal diazepam is often prescribed to be given at the time of recurrence of febrile seizure lasting >5 min (see Table 586-12 for dosing). Alternatively, buccal or intranasal midazolam may be used and is often preferred by parents. Intravenous benzodiazepines, phenobarbital, phenytoin, or valproate may be needed in the case of febrile status epilepticus. If the parents are very anxious concerning their child’s seizures, intermittent oral diazepam can be given during febrile illnesses (0.33 mg/kg every 8 hr during fever) to help reduce the risk of seizures in children known to have had febrile seizures with previous illnesses. Intermittent oral nitrazepam, clobazam, and clonazepam (0.1 mg/kg/day) have also been used. Other therapies have included intermittent diazepam prophylaxis (0.5 mg/kg administered as a rectal suppository every 8 hr), phenobarbital (4-5 mg/kg/day in 1 or 2 divided doses), and valproate (20-30 mg/kg/day in 2 or 3 divided doses). In the vast majority of cases it is not justified to use these medications owing to the risk of side effects and lack of demonstrated long-term benefits, even if the recurrence rate of febrile seizures is expected to be decreased by these drugs. Other antiepileptic drugs (AEDs) have not been shown to be effective.
American Academy of Pediatrics. Febrile seizures: clinical practice guideline for the long-term management of the child with simple febrile seizures. Pediatrics. 2008:1281-1286.
Berkovic SF, Harkin L, McMahon JM, et al. De-novo mutations of the sodium channel gene SCN1A in alleged vaccine encephalopathy: a retrospective study. Lancet Neurol. 2006;5:488-492.
Chen SY, Tsai CN, Lai MW, et al. Norovirus infection as a cause of diarrhea-associated benign infantile seizures. Clin Infect Dis. 2009;48:849-855.
Guerrini R. Epilepsy in children. Lancet. 2006;367:499-524.
Hartfield DS, Tan J, Yager JY, et al. The association between iron deficiency and febrile seizures in childhood. Clin Pediatr. 2009;48:420-426.
Kimia A, Ben-Joseph EP, Rudloe T, et al. Yield of lumbar puncture among children who present with their first complex febrile seizure. Pediatrics. 2010;126:62-69.
Kimia A, Capraro AJ, Hummel D, et al. Utility of lumbar puncture for first simple febrile seizure among children 6 to 18 months of age. Pediatrics. 2009;123:6-12.
Nørgaard M, Ehrenstein V, Mahon BE, et al. Febrile seizures and cognitive function in young adult life: a prevalence study in Danish conscripts. J Pediatr. 2009;155:404-409.
Provenzale JM, Barboriak DP, VanLandingham K, et al. Hippocampus MRI signal hyperintensity after febrile status epilepticus is predictive of subsequent mesial temporal sclerosis. AJR Am J Roentgenol. 2008;190:976-983.
Sadleir LG, Scheffer IE. Febrile seizures. BMJ. 2007;334:307-311.
Strengell T, Uhari M, Tarkka R, et al. Antipyretic agents for preventing recurrences of febrile seizures. Arch Pediatr Adolesc Med. 2009;163:799-804.
Vestergaard M, Pedersen MG, Ostergaard JR, et al. Death in children with febrile seizures: a population-based cohort study. Lancet. 2008;372:457-463.
586.2 Unprovoked Seizures
History and Examination
The history should also include details of the seizure manifestations, particularly those that occurred at its initial onset. These could give clues to the type and brain localization of the seizure. One should question whether there were other previous signs or symptoms that might signify the occurrence of seizures that the parents overlooked or did not report. In some instances, if the events have been going on for a time and there is a question about their nature (e.g., sleep myoclonus versus seizures), then the family can video record the patient and make the video available to the health care provider. Having the parents imitate the seizure can also be helpful. Seizure patterns (e.g., clustering), precipitating conditions (e.g., sleep or sleep deprivation, television, visual patterns, mental activity, stress), exacerbating conditions (e.g., menstrual cycle, medications), frequency, duration, time of occurrence, and other characteristics need to be carefully documented. Parents often overlook, do not report, or underreport absence, complex partial, or myoclonic seizures. A history of personality change or symptoms of increased intracranial pressure can suggest an intracranial tumor. Similarly, a history of cognitive regression can suggest a degenerative or metabolic disease. Certain medications such as stimulants or antihistamines can precipitate seizures. A history of prenatal or perinatal distress or of developmental delay can suggest etiologic congenital or perinatal brain dysfunction. Details of the spells can suggest nonepileptic paroxysmal disorders that mimic seizures (Chapter 587).
Differential Diagnosis
The various types of seizures, as classified by the International League Against Epilepsy (ILAE), are enumerated in Table 586-1. Some seizures might begin with an aura. Auras are sensory experiences reported by the patient and not observed externally. These can take the form of visual (e.g., flashing lights or seeing colors or complex visual hallucinations), somatosensory (tingling), olfactory, auditory, vestibular, or experiential (e.g., déjà vu, déjà vécu feelings) sensations, depending upon the precise localization of the origin of the seizures.
Absence seizures are generalized seizures consisting of staring, unresponsiveness, and eye flutter lasting usually for few seconds. Typical absences are associated with 3 Hz spike–and–slow wave discharges and with petit mal epilepsy, which has a good prognosis. Atypical absences are associated with 1-2 Hz spike–and–slow wave discharges, with head atony and myoclonus during the seizures and with Lennox-Gastaut syndrome, which has a poor prognosis. Seizure type together with the other nonseizure clinical manifestations helps determine the type of epilepsy syndrome with which a particular patient is afflicted (Table 586-7; Chapters 586.3 and 586.4).
Table 586-7 SELECTED EPILEPSY SYNDROMES BY AGE OF ONSET
NEONATAL PERIOD
INFANCY
CHILDHOOD
ADOLESCENCE
AGE-RELATED (AGE OF ONSET LESS SPECIFIC)
SEIZURE DISORDERS THAT ARE NOT TRADITIONALLY GIVEN THE DIAGNOSIS OF EPILEPSY
EPILEPTIC ENCEPHALOPATHIES
OTHER SECONDARY GENERALIZED EPILEPSIES
Lists from International League Against Epilepsy: Table 1: genetic and developmental epilepsy syndromes by age of onset (website). http://www.ilae.org/Visitors/Centre/ctf/CTFtable1.cfm. Accessed October 26, 2010; and International League Against Epilepsy: Table 2. epileptic encephalopathies and other forms of secondary generalized epilepsies (website). http://www.ilae.org/Visitors/Centre/ctf/CTFtable2.cfm. Accessed October 26, 2010.
Guidelines on the evaluation and treatment of a first unprovoked nonfebrile seizure include a careful history and physical examination and brain imaging by head CT or MRI. Emergency head CT in the child presenting with a first unprovoked nonfebrile seizure is often useful for acute management of the patient. Laboratory studies are recommended in specific clinical situations: Spinal tap is considered in patients with suspected meningitis or encephalitis, in children without brain swelling or papilledema, and in children in whom a history of intracranial bleeding is suspected without evidence of such on head CT. In the second of these, examination of the CSF for xanthochromia is essential. CSF tests can also confirm with the appropriate clinical setup the diagnosis of glucose transporter deficiency, cerebral folate deficiency, pyridoxine dependency, pyridoxal dependency, mitochondrial disorders, nonketotic hyperglycemia, and neurotransmitter deficiencies. Electrocardiography (ECG) to rule out long QT or other cardiac dysrhythmias and other tests directed at disorders that could mimic seizures may be needed (Chapter 587).
Approach to the Patient and Additional Testing
The approach to the patient with epilepsy is based on the diagnostic scheme proposed by the ILAE Task Force on Classification and Terminology, presented in Table 586-8. This emphasizes the total approach to the patient, including identification, if possible, of the underlying etiology of the epilepsy and the impairments that result from it. The impairments are very often just as important as, if not more important than, the seizures themselves. There are now many epilepsy syndromes that have been associated with specific gene mutations (see Table 586-2). Different mutations of the same gene can result in different epilepsy syndromes, and mutations of different genes can cause the same epilepsy syndrome phenotype. The clinical use of gene testing in the diagnosis and management of childhood epilepsy has been limited to patients manifesting specific underlying malformational, metabolic, or degenerative disorders, patients with severe named epilepsy syndromes (such as West and Dravet syndromes and progressive myoclonic epilepsies), and, rarely, patients with familial syndromes (see Table 586-2).
Table 586-8 PROPOSED DIAGNOSTIC SCHEME FOR PEOPLE WITH EPILEPTIC SEIZURES AND WITH EPILEPSY
From International League Against Epilepsy: Table 2: proposed diagnostic scheme for people with epileptic seizures and with epilepsy (website). http://www.ilae-epilepsy.org/Visitors/Centre/ctf/table2.cfm. Accessed October 26, 2010.
MR spectroscopy is performed for lactate and creatine peaks.
Deprez L, Jansen A, De Jonghe P. Genetics of epilepsy syndromes starting in the first year of life. Neurology. 2009;72:273-281.
Engel JJr. Report of the ILAE classification core group. Epilepsia. 2006;47:1558-1568.
Guerrini R. Epilepsy in children. Lancet. 2006;367:499-524.
Harden CL, Huff JS, Schwartz TH, et al. Reassessment: neuroimaging in the emergency patient presenting with seizure (an evidence-based review): report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology. 2007;69:1772-1780.
Hirtz D, Ashwal S, Berg A, et al. Practice parameter: evaluating a first nonfebrile seizure in children: report of the quality standards subcommittee of the American Academy of Neurology, the Child Neurology Society, and the American Epilepsy Society. Neurology. 2000;55:616-623.
Krumholz A, Wiebe S, Gronseth G, et al. Practice parameter: evaluating an apparent unprovoked first seizure in adults (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology. 2007;69:1996-2007.
Lagae L. What’s new in genetics in childhood epilepsy. Eur J Pediatr. 2008;167(7):715-722.
586.3 Partial Seizures and Related Epilepsy Syndromes
Secondary Generalized Seizures
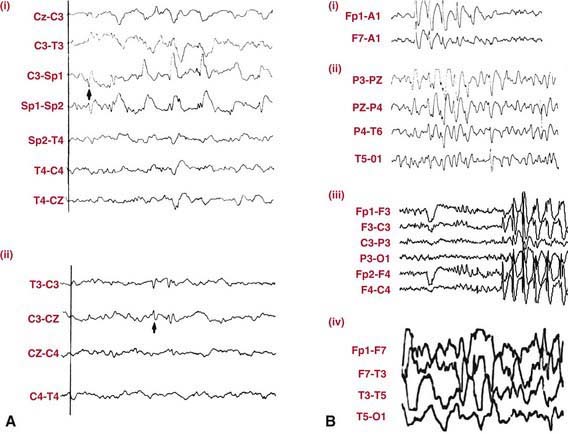
EEG in patients with partial seizures usually shows focal spikes or sharp waves in the lobe where the seizure originates. A sleep-deprived EEG with recording during sleep increases the diagnostic yield and is advisable in all patients whenever possible (Fig. 586-2). Despite that, about 15% of children with epilepsy initially have normal EEGs because the discharges are relatively infrequent or the focus is deep. If repeating the test does not detect paroxysmal findings, then 24-hour video EEG monitoring may be helpful and can allow visualization of the clinical events and the corresponding EEG tracing.
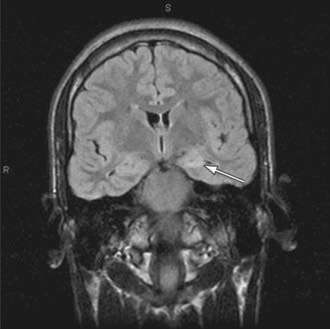
Brain imaging is critical in patients with focal seizures. In general, MRI is preferable to CT and can show pathologies such as changes due to previous strokes or hypoxic injury, malformations, medial temporal sclerosis, arteriovenous malformations, or tumors (Fig. 586-3).
Benign Epilepsy Syndromes with Partial Seizures
In infants, several less-common benign infantile familial convulsion syndromes have been reported. For some of these, the corresponding gene mutation and its function are known (see Tables 586-2 and 586-4), but for others, the genetic underpinnings are yet to be determined. Specific syndromes include benign infantile familial convulsions with parieto-occipital foci linked to chromosomal loci 19q and 2q, benign familial infantile convulsions with associated choreoathetosis linked to chromosomal locus 16p12-q12, and benign infantile familial convulsions with hemiplegic migraine linked to chromosome 1. A number of benign infantile nonfamilial syndromes have been reported, including complex partial seizures with temporal foci, secondary generalized tonic-clonic seizures with variable foci, tonic seizures with midline foci, and partial seizures in association with mild gastroenteritis. All of these have a good prognosis and respond to treatment promptly, often necessitating only short-term (e.g., 6 mo), if any, therapy. Nocturnal autosomal dominant frontal lobe epilepsy has been linked to acetylcholine-receptor gene mutations and manifests with nocturnal seizures with dystonic posturing that respond promptly to carbamazepine. Several other less-frequent familial benign epilepsy syndromes with different localizations have also been described, some of which occur exclusively or predominantly in adults (see Table 586-2).



