Chapter 154 Scleroderma and Raynaud Phenomenon
Classification
Localized scleroderma is distinct from systemic scleroderma and rarely progresses to systemic disease. Within the category of LS there are several subtypes that are differentiated by both the distribution of the lesions and the depth of involvement (Table 154-1). Up to 15% of children have a combination of 2 or more subtypes.
Table 154-1 CLASSIFICATION OF PEDIATRIC SCLERODERMA (MORPHEA)
LOCALIZED SCLERODERMA
Plaque Morphea
Generalized Morphea
Bullous Morphea
Bullous lesions that can occur with any of the subtypes of morphea
Linear Scleroderma
Linear lesions can extend through the dermis, subcutaneous tissue, and muscle to underlying bone; more likely unilateral
Deep Morphea
SYSTEMIC SCLEROSIS
Diffuse
Limited
Clinical Manifestations
Localized Scleroderma
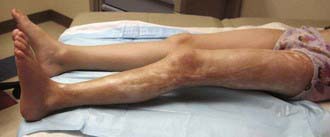
The onset of scleroderma is generally insidious, and manifestations vary according to disease subtype. The initial skin manifestations of localized disease usually include erythema or a bluish hue seen around an area of waxy induration; subtle erythema may be the only presenting sign (Fig. 154-1). Early edema and erythema are followed by indurated, hypopigmented or hyperpigmented, atrophic lesions (Fig. 154-2). Linear scleroderma varies in size from a few centimeters to the entire length of the extremity, with varying depth. Patients sometimes present with arthralgias, synovitis, or flexion contractures (Fig. 154-3). Children also experience limb length discrepancies as a result of growth impairment due to involvement of muscle and bone. Children with en coup de sabre (Fig. 154-4) may have symptoms unique to central nervous system (CNS) involvement, such as seizures, hemifacial atrophy, ipsilateral uveitis, and learning/behavioral changes.
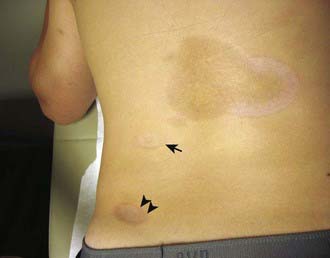
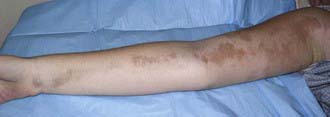
Figure 154-2 Inactive linear scleroderma demonstrating hyperpigmented lesion with areas of normal skin (skip lesions).