Fig. 42.1
Embryonal RMS of the right ear. a Cellular spindled cell proliferation of atypical cells with hyperchromatic nuclei and scant, elongated eosinophilic cytoplasm. The tumor is growing under the keratinizing squamous mucosa seen above the arrowheads. The neoplastic proliferation is more densely cellular under the epithelium and less cellular and myxoid in deeper locations. A densely cellular area at higher magnification is seen in the inset b A large proportion of tumor cells show strong nuclear immunoreactivity for myogenin
Alveolar RMS (20 %): Alveolar RMS is characterized by the presence of fibrovascular septa forming spaces that contain freely floating round malignant cells with abundant eosinophilic cytoplasm (Fig. 42.2). This type is so named because of its distinct architecture resembling that of pulmonary alveoli. While the IRS Pathology Committee originally designated that more than 50 % of the tumor contain these elements to qualify as an alveolar RMS, it is now recognized that any degree of alveolar histology within a tumor specimen is diagnostic [7]. Tumors in the adolescent age group are typically of the alveolar type and have a poorer prognosis because of their aggressive and metastatic nature. Lymphatic and systemic metastases are more common. The prognosis of alveolar tumors is better if a child is less than 10 years of age.
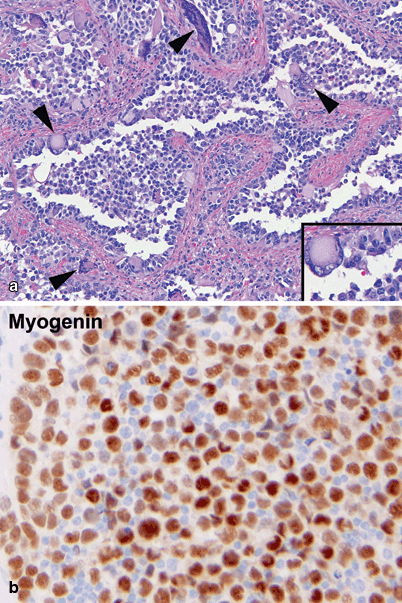
Fig. 42.2
Alveolar rhabdomyosarcoma. a Characteristic alveolar-like spaces delineated by connective tissue trabecules. Tumor cells are large and rounded, have uncohesive growth at the center, and appear to be attached to the connective tissue trabecules at the periphery. Occasional large pleomorphic giant tumor cells are also observed (arrowheads). Higher magnification of rounded tumor cells and of a giant multinucleated tumor cell is shown in the inset. b Most of the tumor cell nuclei are strongly immunoreactive to myogenin
Undifferentiated/Pleomorphic (25 %): Pleomorphic RMS can show features of both embryonal and alveolar histologies [8] while undifferentiated, or RMS “not otherwise specified,” are more difficult to categorize. The vast majority of RMS involving the head and neck, greater than 75 %, fall into the embryonal or alveolar subtypes [9].
Immunohistochemical expression of muscle-specific proteins including desmin, muscle-specific actin, and myogenin are the most important markers for establishing a pathologic diagnosis (Figs. 42.1 and 42.2) [10, 11].
Molecular/Genetic Pathology
RMS are associated with chromosomal abnormalities that can be used to confirm the diagnosis [12]. Some of these abnormalities have important prognostic and possibly therapeutic implications.
Embryonal RMS have characteristically been linked to loss of heterozygosity at a specific site on the short arm of chromosome 11 (11p15). Loss of heterozygosity is associated with over-expression of insulin growth factor 2 and known to play a role in tumorigenesis. Embryonal type tumors have been found to have a DNA content between diploid and hyperdiploid. There have been varying reports as to the prognostic significance of ploidy in RMS, with hyperdiploidy associated with a better long-term survival than tetrapolidy. However, conflicting studies have likewise been published [13–15].
Alveolar RMS has translocations involving the FKHR gene on the long arm of chromosome 13 with genes of the PAX family on either chromosome 2 (PAX3) or chromosome 1 (PAX7) [12]. PAX3 is a transcriptional regulatory protein expressed during embryogenesis and thought crucial for the development of mesenchymal precursors into myoblasts [16]. These translocations result in the fusion of the DNA-binding domain of the neuromuscular developmental transcription factors, encoded by PAX3, to the transcriptional activation domain FKHR (a relatively ubiquitous transcription factor)—the consequences of this fusion are not fully understood [17, 18]. However, the resultant hybrid molecule is a potent transcription activator thought to activate and repress other genes leading to abnormal cell growth.
TP53 mutations have been associated with both embryonal and alveolar histologic subtypes [19, 20]. Expression of this mutated tumor suppressor gene product is associated with decreased survival and anaplastic histology [21, 22].
Elevated n-myc expression (10 % of patients with alveolar), and point mutations in N-ras and K-ras oncogenes (usually embryonal) have been described.
Fluorescent in situ hybridization (FISH) can be used to determine if the above-mentioned translocations are present. It can also be used to assess for break-apart of the FKHR gene. Reverse transcription (i.e. reverse transcription) polymerase chain reaction (RT-PCR) can be helpful in assessing for characteristic translocations seen in alveolar RMS, especially when cytogenetic studies are not possible .
Incidence and Prevalence
In the USA, there are approximately 850 cases of soft tissue sarcoma per year with approximately 40 %, or 350 cases, being RMS. More than 50 % of these cases occur in children younger than 10 years of age [6].
About 35 % of all RMS cases arise in the head and neck region, with nearly 75 % of these tumors confined to the orbit or parameningeal regions [1].
There has been a slight increase in head and neck RMS in the last 30 years with an overall incidence of 0.041 cases per 100,000 population and a statistically significant annual percentage increase of 1.16 % (p < 0.005) [9].
Age Distribution
In a retrospective review of 558 cases of RMS, roughly 2/3rds of head and neck RMS occurred in children 0–19 years of age, with a peak incidence between 0 and 9 years. The remainder occurred in adults aged 20–55 years [9]. The mean age of incidence is 5 years.
Embryonal RMS comprises roughly 80 % of cases in children younger than 5 years of age, and 64 % among 15–19 year olds. The relative percentage of alveolar RMS increases from 15 % among children younger than 5 years of age to 30 % among 15–19 years [6]. This age differentialbetween embryonal and alveolar RMS may partially reflect the higher incidence of head and neck cases in younger children, the majority of which are embryonal .
Sex Predilection
Geographic Distribution
Given that only small case series have been published regarding site-specific experiences with head and neck RMS, data on geographic distribution is not available.
Risk Factors
There is limited evidence that the following environmental exposures are linked to RMS: paternal smoking, maternal use of recreational drugs, antibiotic use, low socioeconomic status, or ionizing radiation (in utero) [24–26]. There is no data linking any of the risks to head and neck RMS in particular.
Relationships to Other Disease States, Syndromes
Most cases of RMS are sporadic; 9 % of cases are syndrome-related. A number of genetic syndromes are thought to be associated with the development of early RMS [27]. (Table 42.1)
Table 42.1
Genetic syndromes associated with pediatric and adolescent cases of rhabdomyosarcoma
1. Neurofibromatosis
2. Beckwith–Weidemann
3. Li Fraumeni syndrome
4. Rubinstein–Taybi syndrome
5. Costello syndrome
6. Gorlin basal cell nevus syndrome
Presentation
Symptoms are dependent upon the tumor’s location at diagnosis. Head and neck RMS is divided into three subtypes: orbital, parameningeal, and non-orbital non-parameningeal [9].
Orbital
Eyelid: this is included in the “orbital category.” Tumors of true eye origin can occur but are rare and arise from the conjunctiva.
Orbit: this region refers to the bony cavity which contains the globe, nerve and vessels, and extraocular muscles. Tumors can occasionally invade the bony walls and extend into the sinuses.
Orbital RMS is most typically associated with the insidious or rapid onset of unilateral proptosis and inferior or inferiotemporal displacement of the globe. Patients can likewise present with worsening eyelid or conjunctival edema and erythema, a palpable mass, ophthalmoplegia (inability to adduct the affected eye), or blepharoptosis (drooping of the upper eyelid). A review by Gandhi et al. reported that approximately 11 % of patients presented after incidental trauma. Only 10–20 % of patients note ocular or orbital pain [29].
Parameningeal (approximately 40 % of head and neck RMS cases)
Middle ear: patients may present with a mass in front or under the ear suggesting a parotid origin. The mass may also extend through the tympanic membrane and appear to be arising from the ear canal. This tumor is often advanced at presentation. Tumors of this type may be associated with otalgia, bloody otorrhea, and hearing loss [30, 31]. They may also be associated with a facial nerve paralysis (VII).
Nasal cavity and paranasal sinuses: patients may present with tumor in any one of the three paranasal sinuses (maxillary, ethmoid, and sphenoid) which surround the nasal cavity. Tumor originating in one sinus will often spread to the others. Tumors arising in the maxillary or ethmoid sinuses may invade the orbit. Patients may have associated rhinosinusitis including nasal obstruction and bloody or mucopurulent rhinorrhea. Occasionally, proptosis can also be present (Fig 42.3).
Nasopharynx: patients present with tumors in the region defined by the back of the nasal septum anteriorly, the sphenoid sinus superiorly, the soft palate inferiorly, and the pharyngeal walls laterally and posteriorly. Tumors of this type may be associated with upper airway obstruction, new onset sleep apnea, or an abducens (VI) nerve palsy.
Infratemporal fossa/pterygopalatine and parapharyngeal areas: patients present with tumors in the region bounded laterally by the medial lobe of the parotid gland and medially by the pharynx.
Approximately 65–80 % of patients with parameningeal disease have high-risk features including intracranial extension, skull base erosion, and cranial nerve paresis at presentation: diplopia caused by involvement of the oculomotor (III), trochlear (IV), and abducens (VI) nerves. A Horner’s syndrome may be the only clinical finding in some instances. Headache and papilledema may be present if there is intracranial involvement. Lymph node involvement is present in 7 % of patients at the time of presentation [10].
A retrospective review by Zorzi et al. of 35 children with parameningeal RMS showed that 54 % presented with a cranial nerve palsy, with the facial nerve being the most commonly involved [32]. In large studies performed by the IRS (Intergroup Rhabdomyosarcoma Study) and European study groups, the nasopharynx and paranasal sinuses are the most commonly reported location of parameningeal tumors [33].
Non-orbital/Non-parameningeal
These tumors arise from both superficial locations (cheek, external ear, scalp), and deep locations (parotid, orophaynx, palate, larynx, and neck).
Superficial tumors may present as a painless mass lesion, while those in deeper locations, such as the oropharynx and larynx, may present with dysphagia, dysphonia, and/or obstructive airway symptoms. Facial nerve paralysis (VII) may be present with tumors of the parotid gland. The majority of non-parameningeal tumors are of the embryonal type, except those occurring in the cheek and scalp which have a tendency to be of the alveolar subtype.
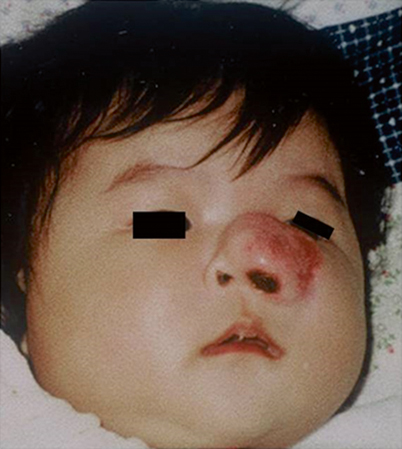
Fig. 42.3
A 4-month old female infant with a rapidly growing lesion in the region of her left nose and maxillary sinus. This lesion was mistakenly diagnosed as an infantile hemangioma, but failed to response to conventional treatment. A biopsy confirmed embryonal rhabdomyosarcoma
Tumors can occur in the thyroid and parathyroid but are rare.
Patterns of Evolution
Parameningeal lesions carry a poorer prognosis given that their deep location renders them clinically silent. They must grow to considerable size before they are visualized from the outside.
The most common sites of metastases for RMS are lung, bone, and bone marrow. Lung metastases are often asymptomatic unless large or progressive whereas bone metastases typically cause pain at the site of the lesion. Bone marrow disease may lead to diminished peripheral blood counts .
Evaluation at Presentation
Physical Exam
Thorough history and physical exam of the head and neck region are recommended to determine the location and size of the mass, and examine for lymphadenopathy. Cranial nerves should be assessed. Flexible nasal endoscopy and laryngoscopy should be routinely performed. Referral to an ophthalmologist, audiologist and ear, nose, and throat (ENT) surgeon are imperative.
Initial Imaging
RMS of the head and neck are best depicted with magnetic resonance imaging (MRI) due to superior soft-tissue contrast, excellent spatial resolution, multiplanar imaging, and absence of ionizing radiation. With these benefits, come the downside of a 30–45 min exam requiring anesthesia for younger children [34].
Routine spin-echo (SE) T1- and T2-weighted sequences before and after intravenous contrast administration are routine. RMS appears relatively hypointense to brain on T2-weighted images. Coronal contrast-enhanced fat-suppressed T1-weighted images are useful for the detection of parameningeal tumors (Fig. 42.4). Volumetric interpolated breath-hold examination (or VIBE) can acquire images rapidly and allow for 3D reconstructions, which can detect subtle skull base abnormalities and perineural tumor spread. Dynamic contrast-enhanced MRI is useful in assessing tumor vascularity in vivo and may be relevant in assessing the nature of a residual mass post-treatment.
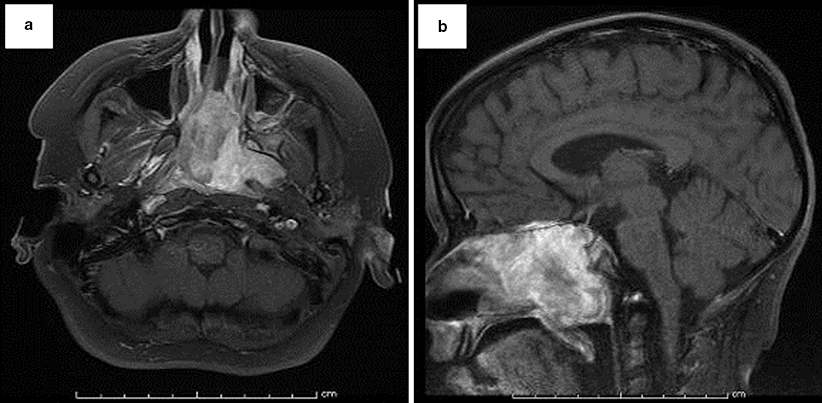
Fig. 42.4
Axial (a) and sagittal (b) T1 weighted high-resolution MR images of the paranasal sinuses of the patient in Fig. 42.3. A large lobulated relatively well-circumscribed heterogeneously enhancing hypercellular mass lesion is present. This lesion is centered within the nasopharyngeal cavity extending into the left inferior orbital fissure, the left masticator space and left pterygopalatine fossa laterally, to the posterior aspect of the nasal air cavity anteriorly, and extends superiorly with invasion of the anterior skull base at the level of the planum sphenoidale and anterior aspect of the pituitary fossa as well as involving the left cavernous sinus with extension superiorly through the left foramen of ovale along V3. Abnormal dural enhancement along the floor of the anterior cranial fossa, left middle cranial fossa, and along the dorsal aspect of the dorsum sella and clivus is also present, suggestive of intracranial extension of the mass lesion
CT scan of the primary site is often performed at diagnosis, but MRI is the preferred imaging modality for follow-up. On CT, RMS appears as a heterogeneous mass, often with necrosis. A CT scan can more readily identify bony erosion [35]. (Fig. 42.5)
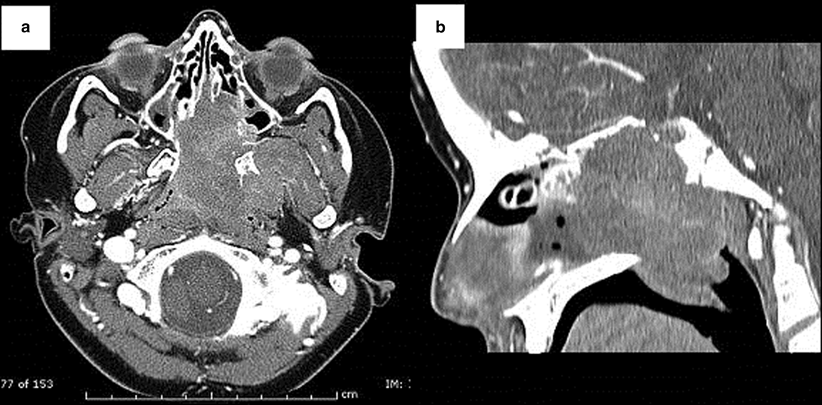
Fig. 42.5
Axial (a) and sagittal (b) CT images of the paranasal sinuses of a 10-year old male who presented with a 6-week history of increasing left nasal obstruction, left otalgia, a hyponasal voice and a 6th cranial nerve palsy. A large nasopharyngeal mass extending into the nasal cavity is present. There is complete destruction of the bony nasal septum and also the anterior wall and floor of the sphenoid sinuses. There is also dehiscence of the bony skull base. Intranasal biopsy confirmed an embryonal rhabdomyosarcoma
Imaging must assess the origin and local extent of the primary tumor, its size in centimeters, extension of tumor beyond the skull base, invasion into the anterior, middle or posterior cranial fossas, dural enhancement, and perineural enhancement [29].
Biopsy
Tissue sampling can occur either via open or needle biopsy. If possible, an open biopsy under anesthesia is preferred. Closed techniques such as fine needle aspiration and tru-cut biopsy are reserved for inaccessible lesions. They obtain smaller volumes of tissue, increase sampling error and inconclusive findings, and often preclude molecular studies [36]. Endoscopic techniques play an important role in exploring and biopsying sinonasal and nasopharyngeal tumors. A frozen section should be obtained at the time of open biopsy to ensure that the specimen is adequate. If it is suggestive of RMS, bone marrow aspiration for cytogenetic analysis can be performed under the same anesthesia.
In the Children’s Oncology Group protocols for RMS, lymph nodes > 15 mm in short axis when assessed by CT scan (slice thickness less than 5 mm) or > 10 mm when palpated clinically, are considered concerning for malignancy. Imaging literature cites that a hypoechoic, rounded lymph node with obliteration of the fat center is likewise concerning [34]. Sampling of concerning lymph nodes by upfront neck dissection is recommended [37].
Stabilization
In most cases, children presenting with head and neck RMS are stable. For patients with orbital RMS who are experiencing vision decline, immediate initiation of chemotherapy is recommended. Steroids to diminish inflammation and topical agents for proptosis are indicated to preserve vision.
Differential Diagnosis
The differential diagnosis of head and neck RMS includes both benign and malignant conditions. The greatest diagnostic challenge in RMS is the distinction from other poorly differentiated small round bluecell tumors (Table 42.2).
Table 42.2
Differential diagnosis for RMS
Oncologic | Benign |
|---|---|
1. Non-Hodgkin’s Lymphoma | Hemangioma/Lymphangioma |
2. Nasopharyngeal carcinoma | Nasal polyp |
3. Osteosarcoma | Schwannoma |
4. Ewing sarcoma and PNET | Plexiform neurofibroma |
5. Juvenile angiofibroma | Giant cell granuloma |
6. Neuroblastoma | Parotid tumor |
7. Fibro-, chondro-, synovial sarcomas | Ossifying fibroma |
8. Lymphadenopathy | Neck cyst |
9. Lymphoproliferative disorders, LCH | Vascular malformation |
10. Optic nerve glioma | Fibrous dysplasia |
Additional Workup/Staging
Laboratory Data
Complete blood count with differential, liver function tests, electrolytes.
May consider LDH, ESR/CRP if considering other etiologies on the differential diagnosis.
Lumbar puncture should be performed for children with orbital or parameningeal tumors with skull base erosion or intracranial extension.
Bilateral bone marrow aspirates and biopsies.
Imaging Evaluation
In addition to imaging of the primary site of disease, diagnostic biopsy, and nodal assessment (as above), a metastatic work-up should be performed as follows:
CT scan of the chest: recommended to assess for pulmonary nodules. This modality is limited by its inability to distinguish benign from malignant lesions [38].
Nuclear medicine bone scan: recommended in European and Children’s Oncology Group protocols, however debate exists as to its yield for osteolytic lesions [39].
Positron Emission Tomography (PET): has been shown to be of equal accuracy in staging the primary tumor, regional lymph nodes and bony metastases [40, 41]. Distant metastases and lymph node metastases have been reported to be superiorly detected by PET/CT over conventional imaging studies [42]. The use of PET in pediatric patients with head and neck RMS is not yet routine, but will likely approach the standard of care.
In order to discuss treatment of head and neck RMS , we must first discuss staging and grouping classifications. The current staging system for treatment protocols was developed by the Soft Tissue Sarcoma Committee of the Children’s Oncology Group (COG-STS) [43].
Staging
Staging is based on the site and size of the tumor and the presence or absence of metastases (Table 42.3). Most tumors of the head and neck present as stage III disease. The 3-year failure-free survival (FFS) rates are reported as 86, 80, 68, and 25 % for Stages I, II, III, and IV in IRS-IV, respectively [43].
Table 42.3
Disease staging dependent on tumor site, size, and the presence or absence of metastases
Stage | Sites | T | Size | N | M |
|---|---|---|---|---|---|
I | Orbit, head and neck (excluding parameningeal) | T1 or T2 | a or b | N0 or N1 or Nx | M0 |
II | Parameningeal
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|