Fig. 37.1
3-year-old boy presenting with left leukocoria
A small proportion of patients with bilateral disease (5–6 %) carry a deletion involving the 13q14 locus, which is large enough to be detected by karyotype analysis. In those cases, retinoblastoma is part of a more complex syndrome resulting from the loss of additional genetic material. Patients with the “13q- syndrome” are characterized by typical facial dysmorphic features, subtle skeletal abnormalities, and different degrees of mental retardation and motor impairment [15]. Dysmorphic features more consistently found include thick anteverted ear lobes, high and broad forehead, prominent philtrum, and short nose. A proportion of patients also have overlapping fingers and toes, microcephaly, and delayed skeletal maturation.
Trilateral retinoblastoma refers to the association of bilateral retinoblastoma with an asynchronous intracranial tumor, which occurs in less than 10 % of bilateral cases [16]. Tumors comprising trilateral retinoblastoma are primitive neuroectodermal tumors (PNETs) exhibiting varying degrees of neuronal or photoreceptor differentiation, suggesting an origin from the germinal layer of primitive cells. The majority of these tumors are pineal region PNETs (pineoblastomas), but in 20–25 % of the cases, the tumors are suprasellar or parasellar. The median age at diagnosis of trilateral retinoblastoma is 23–48 months and the interval between the diagnosis of bilateral retinoblastoma and the diagnosis of the brain tumor is usually more than 20 months [17]. Approximately 5 % of patients with bilateral disease develop pineal cysts; these appear to be a forme fruste of trilateral retinoblastoma [18].
The diagnosis of intraocular retinoblastoma is usually made without pathologic confirmation. An examination under anesthesia with a maximally dilated pupil and scleral indentation is required to examine the entire retina (Fig. 37.2). Endophytic tumors are those that grow inward to the vitreous cavity. Because of its friability, endophytic retinoblastoma may seed the vitreous cavity. Exophytic retinoblastoma grows into the subretinal space, thus causing progressive retinal detachment and subretinal seeding. A very detailed documentation of the number, location, and size of tumors, the presence of retinal detachment and subretinal fluid, and the presence of vitreous and subretinal seeds must be performed. Wide-angle real-time retinal imaging systems such as RetCam® provide a 130° field of view and digital recording, facilitating diagnosis and monitoring.
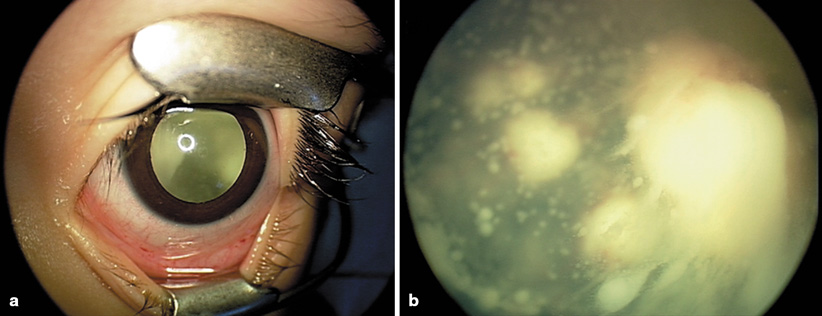
Fig. 37.2
Examination under anesthesia for retinoblastoma. Maximally dilated pupil shows a mass in the posterior chamber (a), closer examination reveals a large endophytic mass with massive vitreous seeding (b)
Additional imaging studies that aid in the diagnosis include bi-dimensional ultrasound, computerized tomography (CT) and magnetic resonance imaging (MRI). These imaging studies are particularly important to evaluate extraocular extension and to differentiate retinoblastoma from other causes of leukocoria (Fig. 37.3). Evaluation for the presence of metastatic disease also needs to be considered in a subgroup of patients. Metastatic disease occurs in approximately 10–15 % of patients, and it usually occurs in association with distinct intraocular histological features, such as deep choroidal and scleral invasion, or with involvement of the iris or ciliary body and optic nerve beyond the lamina cribrosa. In these cases, additional staging procedures, including bone scintigraphy, bone marrow aspirates and biopsies, and lumbar puncture, must be performed .
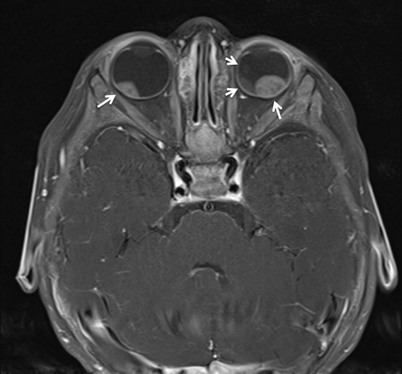
Fig. 37.3
Axial T1 MRI image showing bilateral intraocular masses in an 8-month-old infant with retinoblastoma (arrows)
The Reese-Ellsworth (R-E) grouping system has been generally accepted as the standard for intraocular disease. This grouping system was initially designed to predict the outcome after external beam radiation therapy. It divides eyes into five groups on the basis of the size, location, and number of lesions, and on the presence of vitreous seeding (Table 37.1) [19]. However, developments in the conservative management of intraocular retinoblastoma have made the R-E grouping system less predictable of eye salvage, and less helpful in guiding treatment. A new staging system (International Classification of Retinoblastoma) has been developed, with the goal of providing a simpler, more user-friendly classification more applicable to current therapies. This new system is based on extent of tumor seeding within the vitreous cavity and subretinal space, rather than on tumor size and location, and seems to be a better predictor of treatment success (Fig. 37.4 and Table 37.2) [20].
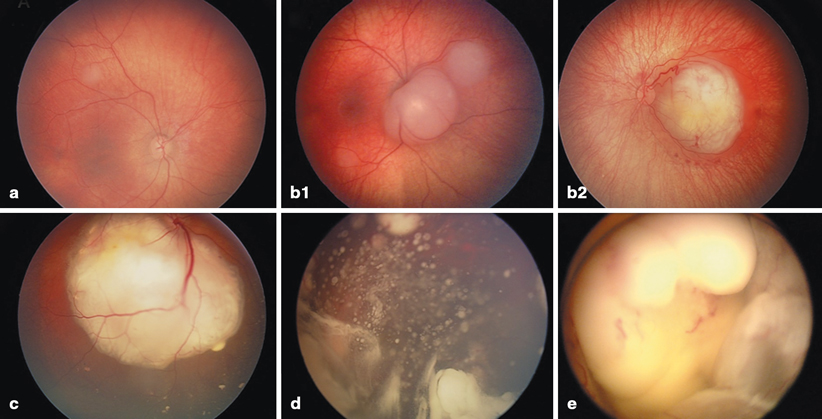
Fig. 37.4
International Classification for Intraocular Retinoblastoma. a Small tumor confined to the retina and distant from the foveola and the optic nerve (Group A). b1 Two small tumors confined to the retina but adjacent to the optic nerve (Group B). b2 Tumor with small amount of subretinal fluid and no subretinal seeding (Group B). c Exophytic retinoblastoma with subretinal fluid and seeding (Group C). d Endophytic retinoblastoma with massive vitreous seeding (Group D). e Large retinoblastoma filling more than two-thirds of the globe (Group E)
Table 37.1
R-E grouping for suitability for treatment of retinoblastoma by radiation therapy
Group I. very favorable | ||
|---|---|---|
Ia | Solitary tumor smaller than 4 dd at or behind the equator | |
Ib | Multiple tumors, none larger than 4 dd, all at or behind equator | |
Group II. favorable | ||
IIa | Solitary tumor 4–10 dd | |
IIb | Multiple tumors 4–10 dd | |
Group III. doubtful | ||
IIIa | Any lesion anterior to equator | |
IIIb | Solitary tumor larger than 10 dd behind equator | |
Group IV. unfavorable | ||
IVa | Multiple tumors, some larger than 10 dd | |
IVb | Any lesion extending anteriorly to the ora serrata | |
Group V. very unfavorable | ||
Va | Massive tumors involving more than half the retina | |
Vb | Vitreous seeding |
Table 37.2
International Classification for Intraocular Retinoblastoma
Group A |
|---|
Small tumors away from foveola and disc |
Tumors ≤ 3 mm in greatest dimension confined to the retina, and |
Located at least 3 mm from the foveola and 1.5 mm from the optic disc |
Group B |
All remaining tumors confined to the retina |
All other tumors confined to the retina not in Group A |
Subretinal fluid (without subretinal seeding) ≤ 3 mm from the base of the tumor |
Group C |
Local subretinal fluid or seeding |
Local subretinal fluid alone > 3 to ≤ 6 mm from the tumor |
Vitreous seeding or subretinal seeding ≤ 3 mm from the tumor |
Group D |
Diffuse subretinal fluid or seeding |
Subretinal fluid alone > 6 mm from the tumor |
Vitreous seeding or subretinal seeding > 3 mm from tumor |
Group E |
Presence of any or more of these poor prognosis features |
More than 2/3 globe filled with tumor |
Tumor in anterior segment |
Tumor in or on the ciliary body |
Iris neovascularization |
Neovascular glaucoma |
Opaque media from hemorrhage |
Tumor necrosis with aseptic orbital cellulitis |
Phthisis bulbi |
For patients undergoing enucleation, pathologic staging that incorporates other features known to influence the modality of treatment and the prognosis, such as choroidal and scleral involvement, optic nerve extension and presence of metastatic disease are used. A newly proposed staging system developed by an international consortium of ophthalmologists and pediatric oncologists incorporates the most important elements of the older systems (Table 37.3) [21]. Growth and invasion occur as a sequence of events, and extraretinal extension occurs only once the tumor has reached large intraocular dimensions. As part of this process, retinoblastoma extends into the ocular coats (choroids and sclera), the optic nerve, and the anterior segment. Extraocular disease is the next step in this progression; locoregional dissemination occurs by direct extension through the sclera into the orbital contents and preauricular lymph nodes, and extraorbital disease manifests as intracranial dissemination and hematogenous metastases .
Table 37.3
International retinoblastoma staging system
Stage 0 | Patients treated conservatively |
|---|---|
Stage I | Eye enucleated, completely resected histologically |
Stage II | Eye enucleated, microscopic residual tumor |
Stage III | Regional extension |
a. Overt orbital disease | |
b. Preauricular or cervical lymph node extension | |
Stage IV | Metastatic disease |
a. Hematogenous metastasis (without central nervous system (CNS) involvement) | |
1. Single lesion | |
2. Multiple lesions | |
b. CNS extension (with or without any other site of regional or metastatic disease) | |
1. Prechiasmatic lesion | |
2. CNS mass | |
3. Leptomeningeal and cerebral spinal fluid (CSF) disease |
Pathology and Pathways of Spread
Macroscopically, retinoblastoma is soft and friable, and it tends to outgrow its blood supply, with resulting necrosis and calcification. Because of its friability, dissemination within the vitreous and retina in the form of small, white nodules (seeds) is common (Fig. 37.2b) [22]. Microscopically, the appearance of retinoblastoma depends on the degree of differentiation. Undifferentiated retinoblastoma is composed of small, round, densely packed cells with hypochromatic nuclei and scant cytoplasm. Several degrees of photoreceptor differentiation have been described and are characterized by distinctive arrangements of tumor cells. The Homer-Wright rosettes are composed of irregular circlets of tumor cells arranged around a tangle of fibrils with no lumen or internal limiting membrane. They are infrequently seen in retinoblastoma and are most often seen in other neuroblastic tumors such as neuroblastoma and medulloblastoma. The Flexner-Wintersteiner rosettes, on the other hand, are specific for retinoblastoma. These structures consist of a cluster of low columnar cells arranged around a central lumen that is bounded by an eosinophilic membrane analogous to the external membrane of the normal retina. These rosettes are seen in 70 % of tumors. The fleurettes are less often seen. In this case, the cells exhibit even more ultrastructural characteristics of photoreceptor differentiation. They are composed of larger cells with abundant eosinophilic cytoplasm arranged in a distinctive fleur-de-lis pattern. Especially well-differentiated tumors composed almost entirely of fleurettes have been called retinomas or retinocytomas. Ultrastructurally, retinoblastoma cells also demonstrate photoreceptor differentiation with the presence of the 9-0 microtubule doublet pattern, abundant cytoplasmic microtubules, synaptic ribbons, and neurosecretory granules. The typical macroscopic and microscopic characteristics of retinoblastoma are depicted in Fig. 37.5 .
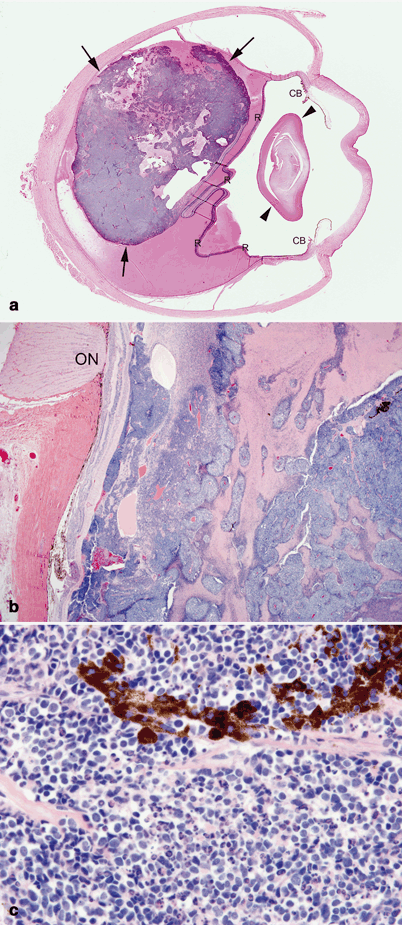
Fig. 37.5
Retinoblastoma. a Cut section of an enucleated eye with posteriorly located retinoblastoma (between arrows) detaching the retina (R). The lens is indicated by arrowheads and the ciliary bodies as CB. b Poorly differentiated retinoblastoma with areas of geographic necrosis. Optic nerve (ON). c Retinoblastoma composed of densely cellular small round cells with numerous apoptotic bodies. A cluster of dark-staining, melanin-laden cells are also observed
Principles of Treatment
Treatment of retinoblastoma aims to save life and preserve vision, and thus needs to be individualized. Factors that need to be considered include unilaterality or bilaterality of the disease, potential for preserving vision, intraocular and extraocular staging [23].
Surgery
Enucleation is indicated for large tumors filling the vitreous for which there is little or no likelihood of restoring vision, and in cases of tumor present in the anterior chamber or in the presence of neovascular glaucoma. Enucleation should be performed by an experienced ophthalmologist; the eye must be removed intact, without seeding the malignancy into the orbit and avoiding globe perforation [24]. For optimal staging, a long section (10–15 mm) of the optic nerve needs to be removed with the globe. An orbital implant is usually fitted during the same procedure, and the extraocular muscles are attached to it. A ceramic prosthetic eye is later fitted in the orbital socket. Orbital exenteration is very seldom indicated. For patients presenting with orbital disease, a judicious use of chemotherapy, surgery (enucleation), and radiation therapy will result in good tumor control, avoiding the need for orbital exenteration .
Focal Therapies
Focal treatments are used for small tumors (less than 3–6 mm), usually in patients with bilateral disease, and in combination with chemotherapy . Photocoagulation with Argon laser is used for the treatment of tumors situated at or posterior to the equator of the eye, and for the treatment of retinal neovascularization due to radiation therapy [25]. This technique is limited to tumors measuring no greater than 4.5 mm in base, and no greater than 2.5 mm in thickness. The treatment is directed to coagulate all blood supply to the tumor. Cryotherapy is used for the treatment of small equatorial and peripheral lesions, measuring no more than 3.5 mm in base and no more than 2 mm thickness [26]. One or two monthly sessions of triple freeze and thaw are performed, and tumor control rates are usually excellent. Finally, an important focal method is transpupillary thermotherapy, which applies focused heat at subphotocoagulation levels, usually with diode laser [27]. In thermotherapy, the goal is to deliver a temperature of 42–60 ° C for 5–20 min to the tumor, sparing retinal vessels from photocoagulation. The use of focal treatments is especially important in conjunction with chemotherapy, and both treatment modalities appear to have a synergistic effect. In general, local control rates of 70–80 % can be achieved. Complications of focal treatments include transient serous retinal detachment, retinal traction and tears, and localized fibrosis .
Chemotherapy
Chemotherapy is indicated in patients with extraocular disease, in the subgroup of patients with intraocular disease with high-risk histological features, and in patients with bilateral disease in conjunction with aggressive focal therapies. Agents effective in the treatment of retinoblastoma include platinum compounds, etoposide, cyclophosphamide, doxorubicin, vincristine, and ifosfamide [23].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


