Arasen A. V. Paupoo

Puberty is the period during which secondary sexual characteristics develop and the capability of sexual reproduction is attained. The physical changes accompanying pubertal development result directly or indirectly from maturation of the hypothalamus, stimulation of the sex organs, and secretion of sex steroids. Hormonally, puberty in humans is characterized by the resetting of the classic negative gonadal steroid feedback loop, alterations in circadian and ultradian (frequent) gonadotropin rhythms, and the acquisition in the woman of a positive estrogen feedback loop, which controls the monthly rhythm as an interdependent expression of gonadotropins and ovarian steroids.
The ability to evaluate and treat aberrations of pubertal development requires an understanding of the normal hormonal and physical changes that occur at puberty. An understanding of these changes is important in evaluating young women with amenorrhea.
Normal Pubertal Development
Factors Affecting Time of Onset
The major determinant of the timing of the onset of puberty is no doubt genetic, but a number of other factors appear to influence both the age at onset and the progression of pubertal development. Among these influences are nutritional state, general health, geographic location, exposure to light, and psychological state (1). The concordance of the age of menarche in mother–daughter pairs and between sisters and in ethnic populations illustrates the importance of genetic factors (1). Typically, the age of menarche is earlier than average in children with moderate obesity (up to 30% above normal weight for age), whereas delayed menarche is common in those with severe malnutrition. Children who live in urban settings, closer to the equator, and at lower altitudes typically begin puberty earlier than those who live in rural areas, farther from the equator, and at higher elevations. The risk of earlier onset of puberty is 10 to 20 times greater after international adoption for unclear reasons (2). Other risk factors implicated for precocious puberty include exposure to estrogenic endocrine-disrupting chemicals and the absence of a father in the home (3,4). Blind girls apparently undergo menarche earlier than sighted girls, suggesting some influence of light (5).
In Western Europe, the age of menarche declined 4 months each decade between 1850 and 1960 (1). Data suggest that the trend toward earlier pubertal development may be continuing among girls (but not boys) who live in the United States (6). It is presumed that these changes represent improved nutritional status and healthier living conditions.
One of the more controversial hypotheses centers on the role of total body weight and body composition on the age of menarche. It is argued that a girl must reach a critical body weight (47.8 kg) before menarche can occur (7). Body fat must increase to 23.5% from the typical 16% of the prepubertal state, which presumably is influenced by nutritional status (8). This hypothesis is supported by observations that menarche occurs earliest in obese girls, followed by normal-weight girls, then underweight girls, and lastly anorectic girls (Fig. 29.1). The importance of other factors is indicated by observations that menarche is often delayed in morbidly obese girls, those with diabetes, and those who exercise intensely but are of normal body weight and body fat percentage. Girls with precocious puberty may undergo menarche even if they have a low body fat percentage, and other girls show no pubertal development with a body fat percentage of 27% (9). The hypothesis linking menarche to body weight and composition does not always seem valid because menarche is a late event in pubertal development.
Figure 29.1 Normal twins at 12 years of age. The heavier twin (weighing 143 lb) is clearly more advanced in puberty than the lighter twin (weighing 87 lb). Anecdotal photographs and data such as these served to provide the basis for the theory that body fat, body mass, and menarche are linked. (From Wilkins L. The diagnosis and treatment of endocrine disorders in childhood and adolescence. 3rd ed. Springfield, IL: Charles C Thomas, 1965, with permission.)
Physical Changes during Puberty
The changes associated with puberty occur in an orderly sequence over a definite time frame. Any deviation from this sequence or time frame should be regarded as abnormal. The pubertal changes, their relationship to one another, and the ages at which they occur are distinctly different in girls than in boys. Although this chapter focuses on girls, changes in boys are considered briefly.
Tanner Stage
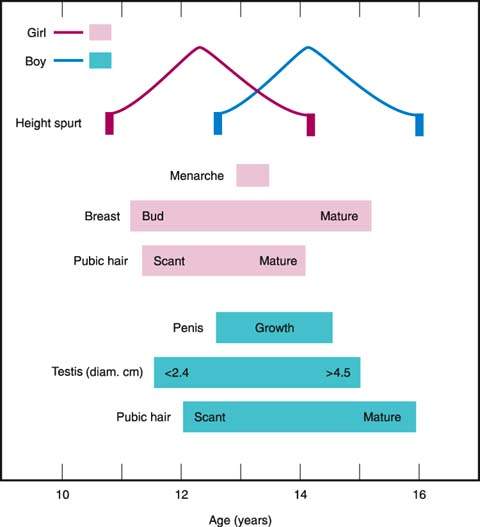
In girls, pubertal development typically takes place over 4.5 years (Fig. 29.2). The first sign of puberty is accelerated growth, and breast budding is usually the first recognized pubertal change, followed by the appearance of pubic hair, peak growth velocity, and menarche. The stages initially described by Marshall and Tanner are often used to describe breast and pubic hair development (10).
Figure 29.2 Schematic sequence of events at puberty. An idealized average girl and an idealized average boy are represented. (From Rebar RW. Practical evaluation of hormonal status. In: Yen SSC, Jaffe RB, eds. Reproductive endocrinology: physiology, pathophysiology and clinical management. 3rd ed. Philadelphia, PA: WB Saunders, 1991:830, with permission; based on data from Marshall WA, Tanner JM. Variations in patterns of pubertal changes in girls. Arch Dis Child 1969;44:291–303, and Marshall WA, Tanner JM. Variation in the pattern of pubertal changes in boys. Arch Dis Child 1970;45:13–23, with permission.)
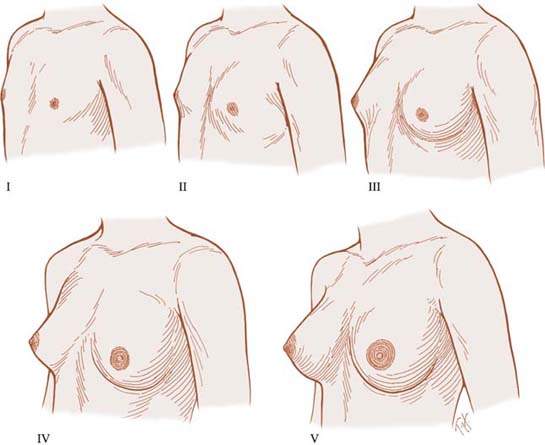
With regard to breast development (Fig. 29.3), Tanner stage 1 refers to the prepubertal state and includes no palpable breast tissue, with the areolae generally less than 2 cm in diameter. The nipples may be inverted, flat, or raised. In Tanner stage 2, breast budding occurs, with a visible and palpable mound of breast tissue. The areolae begin to enlarge, the skin of the areolae thins, and the nipple develops to varying degrees. Tanner stage 3 is reflected by further growth and elevation of the entire breast. When the individual is seated and viewed from the side, the nipple is generally at or above the midplane of breast tissue. In most girls, Tanner stage 4 is defined by projection of the areola and papilla above the general breast contour in a secondary mound. Breast development is incomplete until Tanner stage 5, in which the breast is mature in contour and proportion. In most women, the nipple is more pigmented at this stage than earlier in development, and Montgomery’s glands are visible around the circumference of the areola. The nipple is generally below the midplane of breast tissue when the woman is seated and viewed from the side. Full breast development usually occurs over 3 to 3.5 years, but it may occur in as little as 2 years or not progress beyond stage 4 until the first pregnancy. Breast size is no indication of breast maturity.
Figure 29.3 Diagrammatic depiction of Tanner breast stages in adolescent women. (From Ross GT, Van de Wiele RL, Frantz AG. The ovaries and the breasts. In: Williams RH, ed. Textbook of endocrinology. 6th edition. Philadelphia, PA: WB Saunders, 1981:355, with permission; adapted from Marshall WA, Tanner JM. Variations in patterns of pubertal changes in girls. Arch Dis Child 1969;44:291–303).
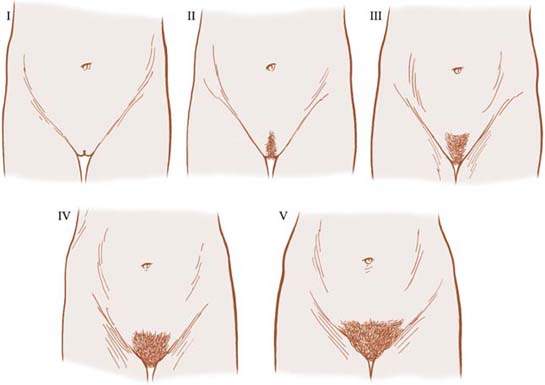
Pubic hair staging is related both to quantity and distribution (Fig. 29.4). In Tanner stage 1, there is no sexually stimulated pubic hair present, but some nonsexual hair may be present in the genital area. Tanner stage 2 is characterized by the first appearance of coarse, long, crinkly pubic hair along the labia majora. In Tanner stage 3, coarse, curly hair extends onto the mons pubis. Tanner stage 4 is characterized by adult hair in thickness and texture, but the hair is not distributed as widely as in adults and typically does not extend onto the inner aspects of the thighs. Except in certain ethnic groups, including Asians and American Indians, pubic hair extends onto the thighs in Tanner stage 5.
Figure 29.4 Diagrammatic depiction of Tanner pubic hair staging in adolescent women. (From Ross GT, VandeWiele RL, Frantz AG. The ovaries and the breasts. In: Williams RH, ed. Textbook of endocrinology. 6th ed. Philadelphia, PA: WB Saunders, 1981:355, with permission; adapted from Marshall WA, Tanner JM. Variations in patterns of pubertal changes in girls. Arch Dis Child 1969;44:291–303.)
The staging of male pubertal sexual maturation is based on genital size and pubic hair development. Tanner stage 1 is prepubertal. Tanner stage 2 of genital growth begins when testicular enlargement is first evident. Testis length along the longitudinal axis ranges from 2.5 to 3.2 cm. The size of the penis increases. Pigmented, curly pubic hair is first visible around the base of the penis. In Tanner stage 3, there is further growth of the penis in both length and diameter, the scrotum develops further, and testis length increases to 3.3 to 4 cm. Thicker, curly hair extends above the penis. Tanner stage 4 involves further growth of the genitalia, with testis length ranging from 4 to 4.5 cm. Extension of pubic hair over the genital area continues, but the volume is less than in the adult. At this stage, the prostate gland is palpable by rectal examination. In Tanner stage 5, the genitalia are within the adult range in size. Average flaccid penile length in adult men ranges between 8.6 and 10.5 cm from tip to base. Pubic hair spreads laterally onto the medial thighs. Hair may or may not extend from the pubic area toward the umbilicus and anus.
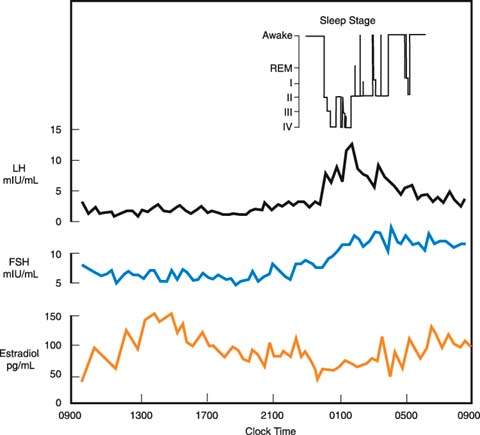
Figure 29.5 Patterns of circulating luteinizing hormone (LH), follicle-stimulating hormone (FSH), and estradiol in a stage 3 pubertal girl over a 24-hour period with the encephalographic stage of sleep indicated. (From Boyar RM, Wu RHK, Roffwarg H, et al. Human puberty: 24-hour estradiol patterns in pubertal girls. J Clin Endocrinol Metab 1976;43:1418–1421, with permission.)
Pigmented pubic hair is often the first recognized sign of male puberty even though it typically occurs 6 months after genital growth begins. Tanner stage 3 puberty often is accompanied by symmetric or asymmetric gynecomastia, and mature sperm first can be identified with microscopic urinalysis.
Height and Growth Rate
Plotting height increments (i.e., growth velocity) against the phases of puberty allows one to see relationships during puberty (Fig. 29.2). Girls reach peak height velocity early in puberty before menarche. As a consequence, they have limited growth potential after menarche. In contrast, boys reach peak height velocity about 2 years later than girls. Boys grow an average of 28 cm during the growth spurt, in comparison to a mean of 25 cm for girls. Adult men eventually are an average of 13 cm taller than adult women because they are taller at the onset of the growth spurt. Hormonal control of the pubertal growth spurt is complex. Growth hormone (GH), insulinlike growth factor 1 (IGF-1), and gonadal steroids play major roles. Adrenal androgens appear to be less important. Mutations limiting conversion of androgens to estrogens in males confirmed that estrogen is the major stimulus to the pubertal growth spurt in both boys and girls (11).
During the growth spurt associated with puberty, the long bones in the body lengthen and the epiphyses ultimately close. The bone or skeletal age of any individual can be estimated closely by comparing x-rays documenting the development of bones in the nondominant hand (most commonly), knee, or elbow to standards of maturation for the normal population. The Greulich and Pyle atlas is used most often for this purpose (12). Skeletal age is more closely correlated with pubertal stage than with chronologic age during puberty. With height and chronologic age, an individual’s bone age can be used to predict final adult height using the Bayley-Pinneau tables (13). Bone age determinations can be used to assess the degree of delay, monitor subsequent development, and estimate final adult height.
Another practical clinical approach to predicting adult height uses midparental height. The adjusted midparental height is calculated by adding 13 cm to the mother’s height (for boys) or subtracting 13 cm from the father’s height (for girls) and then determining the mean of the heights of the parents, including the adjusted height of the opposite-sex parent. Adding and subtracting 8.5 cm to the calculated predicted height approximates the target range of the 3rd to the 97th percentile for the anticipated adult height of the child. This quick calculation can be of assistance in evaluating individuals with delayed or precocious pubertal development and those with short stature.
Several changes in body composition occur during pubertal development. Although lean body mass, skeletal mass, and body fat are equal in prepubertal boys and girls, by maturity, men have 1.5 times the lean body mass and almost 1.5 times the skeletal mass of women, whereas women have twice as much body fat as men (1). The changes in body contour in girls, with accumulation of fat at the thighs, hips, and buttocks, occur during the pubertal growth spurt. In this regard, testosterone is a potent anabolic steroid and is responsible for the major changes in boys, whereas estrogen increases total body fat in a characteristic distribution at the thighs, buttocks, and abdomen in girls.
Other physical changes show sexual dimorphism at puberty. In boys both the membranous and cartilaginous portions of the vocal cords lengthen much more than they do in girls, accounting for deepening of the voice. Comedones, acne, and seborrhea of the scalp begin because of increased secretion of adrenal and gonadal steroids at puberty. In general, early-onset acne correlates with the development of severe acne later in puberty. The appearance of comedones in the nasal creases and behind the pinna may be the first indications of impending pubertal development.
Hormonal Changes
By 10 weeks of gestation, gonadotropin-releasing hormone (GnRH) is present in the hypothalamus, and luteinizing hormone (LH) and follicle-stimulating hormone (FSH) are present in the pituitary gland (14). Gonadotropin levels are elevated in both female and male fetuses before birth; the levels of FSH are higher in females. At birth, gonadotropin and sex steroid concentrations are still high, but the levels decline during the first several weeks of life and remain low during the prepubertal years. The hypothalamic–pituitary unit appears to be suppressed by the extremely low levels of gonadal steroids present in childhood. Gonadal suppression of gonadotropin secretion is demonstrated by higher gonadotropin levels in children with gonadal dysgenesis and those who undergo gonadectomy before puberty (15).
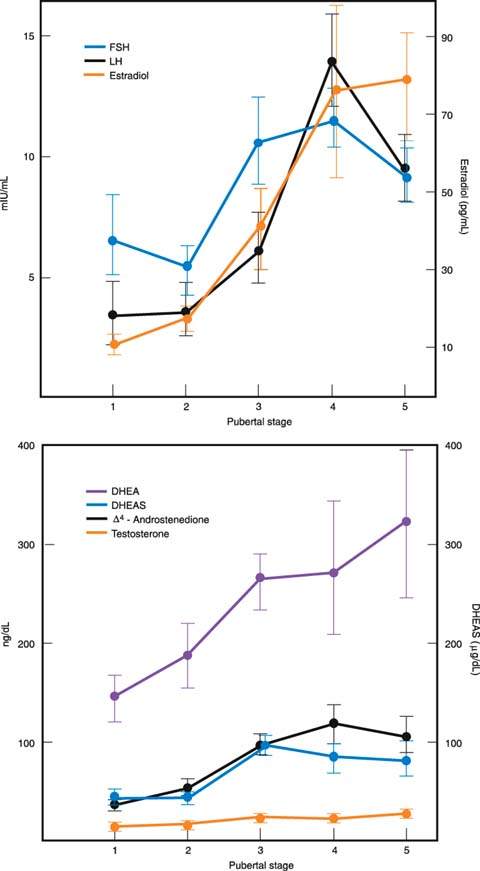
Several of the hormonal changes associated with pubertal development begin before any of the physical changes are obvious. Early in puberty, there is increased sensitivity of LH to GnRH. Sleep-entrained increases in both LH and FSH can be documented early in puberty (16). In boys, the nocturnal increases in gonadotropin levels are accompanied by simultaneous increases in circulating testosterone levels (17). In contrast, in girls, the nighttime increases in circulating gonadotropin levels are followed by increased secretion of estradiol the next day (18) (Fig. 29.5). This delay in estradiol secretion is believed to result from the additional synthetic steps required in the aromatization of estrogens from androgens. Basal levels of both FSH and LH increase through puberty. The patterns differ in boys and girls, with LH levels (measured in mIU/mL) eventually becoming greater than FSH levels (19) (Fig. 29.6). Although it now appears that gonadotropins are always secreted in an episodic or pulsatile fashion, even before puberty, the pulsatile secretion of gonadotropins is more easily documented as puberty progresses and basal levels increase (20).
Increased adrenal androgen secretion is important in stimulating adrenarche, the appearance of pubic and axillary hair, in both boys and girls. Pubarche specifically refers to the appearance of pubic hair. Progressive increases in circulating levels of the major adrenal androgens, dehydroepiandrosterone (DHEA) and its sulfate (DHEAS), begin as early as 2 years of age, accelerate at 7 to 8 years of age, and continue until 13 to 15 years of age (21–23). The accelerated increases in adrenal androgens begin about 2 years before the increases in gonadotropin and gonadal sex steroid secretion when the hypothalamic–pituitary–gonadal unit is still functioning at a low prepubertal level.
In girls, mean levels of estradiol, secreted predominantly by the ovaries, increases steadily during puberty (19). Although, as noted, increases in estradiol first appear during the daytime hours, basal levels eventually increase during both the day and night. Estrone, which is secreted in part by the ovaries and arises in part from extraglandular conversion of estradiol and androstenedione, increases early in puberty but plateaus by midpuberty. Thus, the ratio of estrone to estradiol decreases throughout puberty, indicating that ovarian production of estradiol becomes increasingly important and peripheral conversion of androgens to estrone becomes less important during maturation.
In boys, most of the testosterone in the circulation arises from direct secretion by the Leydig cells of the testis. Testosterone induces development of a male body habitus and voice change, whereas dihydrotestosterone (DHT), produced following 5α reduction within target cells, induces enlargement of the penis and prostate gland, beard growth, and temporal hair recession during puberty. Mean plasma testosterone levels rise progressively during puberty, with the greatest increase occurring during Tanner stage 2 (24).
Growth hormone secretion increases along with increased gonadotropin secretion at the onset of puberty. It is believed that the increase in GH is mediated by estrogen, which in boys is dependent on aromatization of testosterone to estradiol and reflects increasing sex steroid production at puberty. Nonetheless, there are profound sex differences in GH secretion during puberty. Girls have higher basal levels of GH throughout puberty, reaching maximal levels around the time of menarche and decreasing thereafter. In contrast, basal concentrations of GH remain constant throughout puberty in boys. Growth hormone secretion is highly pulsatile, with most pulses occurring during sleep and with sex steroids increasing pulse amplitude rather than altering pulse frequency.
Growth hormone stimulates production of IGF-1 in all tissues, with concentrations found in the circulation spilling over from the liver. During puberty the negative feedback effect of IGF-1 on GH secretion must be reduced because both IGF-1 and GH levels are high. GH and IGF-1 play significant roles in the changes in body composition that occur at puberty because both hormones are potent anabolic agents.
In the final stages of puberty in both boys and girls, GH secretion begins to diminish, returning to prepubertal levels in adult life, despite continued exposure to high levels of gonadal steroids.
Mechanisms Underlying Puberty
The mechanisms responsible for the numerous hormonal changes that occur during puberty are poorly understood, although it is recognized that a “central nervous system program” must be responsible for initiating puberty. It appears that the hypothalamic–pituitary–gonadal axis in girls develops in two distinct stages during puberty. First, sensitivity to the negative or inhibitory effects of the low levels of circulating sex steroids present in childhood decreases early in puberty. Second, late in puberty, there is maturation of the positive or stimulatory feedback response to estrogen, which is responsible for the ovulatory midcycle surge of LH.
Current evidence suggests that the central nervous system inhibits the onset of puberty until the appropriate time (25). Based on this theory, the neuroendocrine control of puberty is mediated by GnRH-secreting neurons in the medial basal hypothalamus, which together act as an endogenous pulse generator. At puberty, the GnRH pulse generator is reactivated (i.e., disinhibited), leading to increased amplitude and frequency of GnRH pulses. In turn, the increased GnRH secretion results in increased gonadotropin and then gonadal steroid secretion. What causes this “disinhibition” of GnRH release is unknown.
Figure 29.6 Increases (± standard error) in circulating levels of gonadotropins and adrenal and gonadal steroids through puberty in girls. DHEA, dehydroepiandrosterone; DHEAS, dehydroepiandrosterone sulfate. (From Emans SJH, Goldstein DP. The physiology of puberty. In: Emans SJH, Goldstein DP, eds. Pediatric and adolescent gynecology. 3rd ed. Boston, MA: Little, Brown, 1990:95, with permission.)
The relationship between body mass and the onset of puberty focused attention on leptin, produced by adipocytes, as a candidate for the factor initiating puberty. In the infertile leptin-deficient mouse, leptin therapy can induce sexual maturation and maintain fertility. Observations of two patients with leptin receptor mutations who failed to enter puberty suggest that leptin may have a similar role in humans (25).
Longitudinal studies of leptin secretion noted that there is increased leptin secretion around the time of pubertal onset. Leptin levels are increased throughout puberty in girls but not in boys. There is speculation that leptin is a trigger for pubertal onset, but a more widely held view is that leptin plays a more permissive role in regulating pubertal onset (25).
Aberrations of Pubertal Development
Classification
Several aberrations of pubertal development, as detailed in Table 29.1, can occur in girls. Pubertal aberrations can be classified in four broad categories:
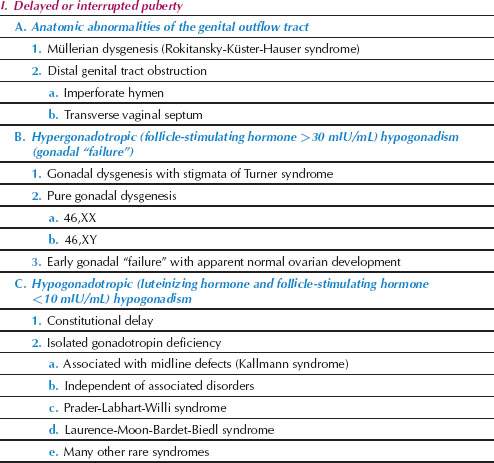

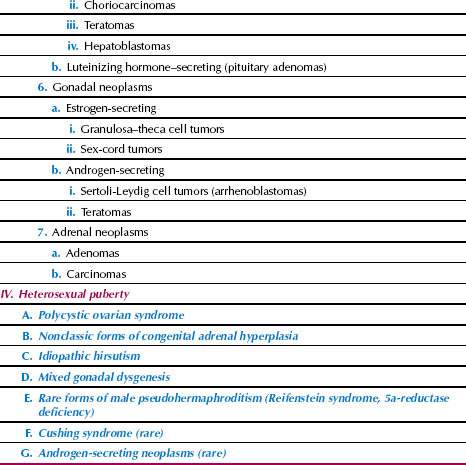
Disorders of sexual development and amenorrhea may be considered in relation to this classification of the aberrations of puberty. It is very helpful to document the growth of the individual and to plot the individual’s height and weight on one of several commonly available growth charts (Fig. 29.7).
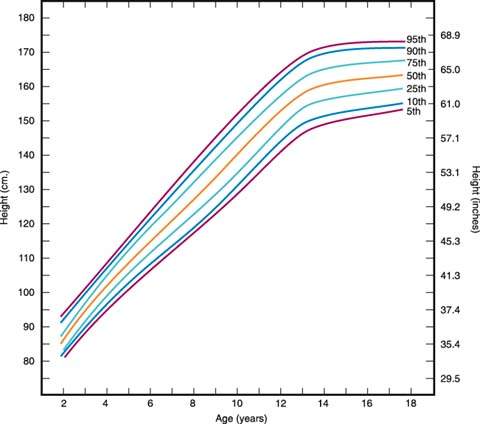
Figure 29.7 Growth chart showing stature by age percentiles for girls aged 2 to 18 years. Weight can be plotted in a similar fashion. Several excellent growth charts are available to clinicians, including those from Ross Laboratories (Columbus, OH), Serono Laboratories (Randolph, MA), and Genentech, Inc. (South San Francisco, CA). (From Hamill PVV, Drizd TA, Johnson CL, et al. Physical growth: National Center for Health Statistics percentiles. Am J Clin Nutr 1979;32:607–629, with permission; based on data from the National Center for Health Statistics.)
Delayed or Interrupted Puberty
The history and physical examination, with particular attention to growth, are most important in the evaluation of individuals with delayed puberty. Pubertal delay is much more common in boys than in girls. It is important to remember that puberty may be delayed in any child suffering from any severe chronic disease, including celiac disease, Crohn disease, sickle cell anemia, and cystic fibrosis. Chronic illness should be reviewed during the history and physical examination. One possible approach to evaluation is depicted in Figure 29.8.
Figure 29.8 Flow diagram for the evaluation of delayed or interrupted pubertal development, including primary amenorrhea, in phenotypic girls. Girls with asynchronous development often present because of failure to menstruate. FSH, follicle-stimulating hormone; PRL, prolactin; T4, thyroxine; TSH, thyroid-stimulating hormone; CNS, central nervous system; MRI, magnetic resonance imaging; CT, computed tomography. (From Rebar RW. Normal and abnormal sexual differentiation and pubertal development. In: Moore TR, Reiter RC, Rebar RW, et al., eds. Gynecology and obstetrics: a longitudinal approach. New York: Churchill Livingstone, 1993:97–133, with permission.)
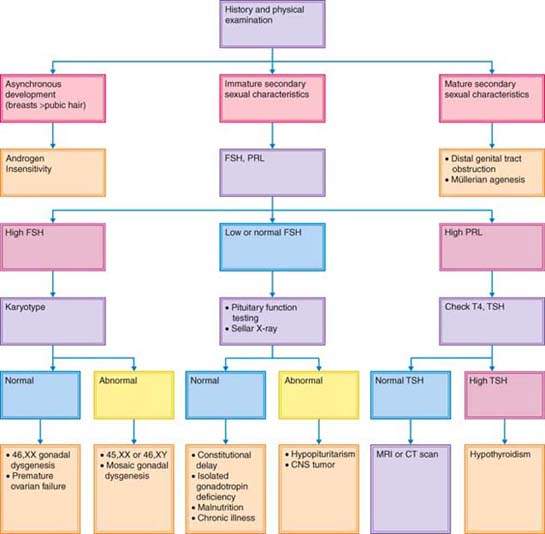
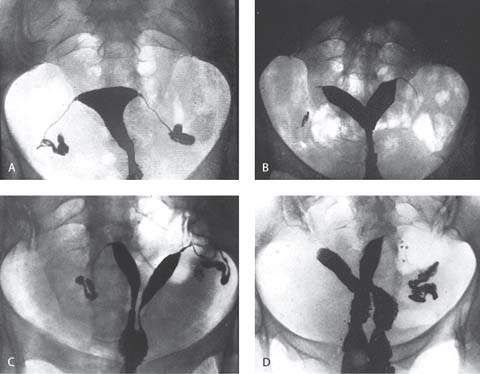
Figure 29.9 Hysterosalpingograms of normal and abnormal female genital tracts. The radiographic photographs have been reversed to accentuate the uterine cavities. A: Normal study with bilateral spill. B: Bicornuate uterus. C: Uterus didelphis. D: Uterus didelphis with double vagina. (Courtesy of Dr. A. Gerbie; from Spitzer IB, Rebar RW. Counselling for women with medical problems: ovary and reproductive organs. In: Hollingsworth D, Resnik R, eds. Medical counselling before pregnancy. New York: Churchill Livingstone, 1988:213–248, with permission.)
Anatomic Abnormalities of the Genital Outflow Tract
Those girls who have mature secondary sex characteristics and any of a number of disorders of the outflow tract and uterus, often termed müllerian agenesis and dysgenesis, are most often identified on examination (Fig. 29.9). One of the most logical classification schemes that was proposed is shown in Table 29.2 (27). The incidence of these anomalies was estimated to be 0.02% of the female population several, but the incidence may have increased as a result of the maternal ingestion of diethylstilbestrol (DES) and the resultant increase in anomalies of the lumen of the uterus (class VI) (28,29). Of the disorders unrelated to drug use, the septate uterus (class V) is most common.
Disorders of the outflow tract and uterus often occur as a part of a syndrome of malformations that include abnormalities of the skeletal and renal systems (Mayer-Rokitansky-Küster-Hauser syndrome). Familial aggregates of the most common disorders of müllerian differentiation in girls—müllerian aplasia and incomplete müllerian fusion—are best explained on the basis of polygenic and multifactorial inheritance (30). It is clear that the HOX genes, a family of regulatory genes that encode for transcription factors, are essential for proper development of the müllerian tract in the embryonic period, and HOXA 13 is altered in hand–foot–genital syndrome (31). WNT4 may be involved in uterine development, as a WNT4 mutation was described in cases involving a Mayer-Rokitansky-Küster-Hauser-like syndrome with hyperandrogenism (32).
The most common single anatomic disorder of puberty is the imperforate hymen, which prevents the passage of endometrial tissue and blood. These products can accumulate in the vagina (hydrocolpos) or uterus (hydrometrocolpos) and result in a bulging hymen that is often bluish in color. The affected individual often has a history of vague abdominal pain with approximately monthly exacerbations. It is sometimes difficult to distinguish an imperforate hymen from a transverse vaginal septum, and in most situations, examination under anesthesia is required.
Regardless of the cause, uterine anomalies not involving segmental müllerian agenesis or hypoplasia (class I) are compatible with normal pregnancy. However, increased fetal wastage is reported in the presence of these anomalies (33). Uterine malformations are associated with spontaneous abortion, preterm labor, abnormal presentations, and complications of labor (i.e., retained placenta). Many of these uterine anomalies can be identified with hysterosalpingography (Fig. 29.9). Hysterosalpingography, laparoscopy, and hysteroscopy are used to differentiate a septate uterus (class V) from a bicornuate uterus (class IV). Magnetic resonance imaging (MRI) and endovaginal ultrasonography (sometimes with sonohysterography) are as accurate as these invasive techniques in identifying the abnormality (34).
Obstruction or malformation of the distal genital tract must be distinguished from androgen insensitivity. Individuals with androgen insensitivity have breast development in the absence of significant pubic and axillary hair development; the vagina may be absent or foreshortened in these women.
Hypergonadotropic and Hypogonadotropic Hypogonadism
Basal levels of FSH and prolactin should be determined in individuals in whom secondary sex characteristics have not developed to maturity (Fig. 29.8). Bone age should be estimated from x-rays of the nondominant hand. If prolactin levels are elevated, thyroid function should be assessed to determine whether the individual has primary hypothyroidism. Paradoxically, primary hypothyroidism can result in precocious puberty. If thyroid function is normal, a hypothalamic or pituitary neoplasm is possible, and careful evaluation of the hypothalamic and pituitary area by MRI or computed tomography (CT) is indicated.
The karyotype should be determined in any individual with delayed puberty and increased basal FSH concentrations. Regardless of the karyotype, the individual with hypergonadotropic hypogonadism has some form of ovarian “failure” (i.e., primary hypogonadism).
Forms of Gonadal Failure
Turner Syndrome
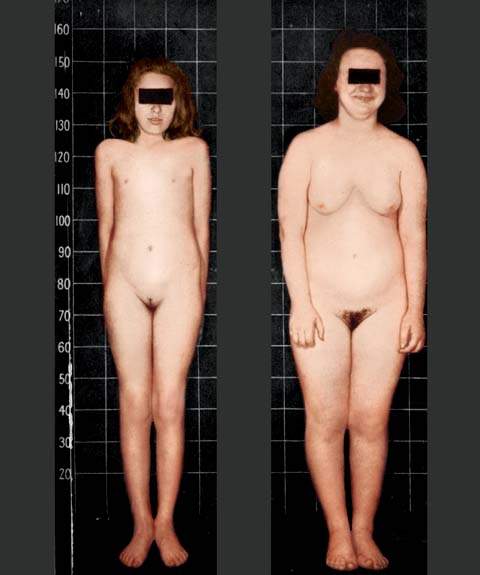
The diagnosis of Turner syndrome requires the presence of characteristic features in phenotypic females coupled with complete or partial absence of the second sex chromosome, with or without cell line mosaicism. Most affected individuals have a 45,X karyotype, while others have mosaic karyotypes (i.e., 45,X/46,XX; 45,X/46,XY). Intrauterine growth restriction is common in infants with a 45,X karyotype. After birth, these patients generally grow slowly, beginning in the second or third year of life. They typically have many of the associated stigmata, including lymphedema and sometimes large cystic hygromas of the neck at birth; a webbed neck; multiple pigmented nevi; disorders of the heart, kidneys (most commonly horseshoe), and great vessels (most commonly coarctation of the aorta); and small hyperconvex fingernails (35) (Fig. 29.10). Diabetes mellitus, thyroid disorders, essential hypertension, and other autoimmune disorders are often present in individuals with 45,X karyotypes.
Figure 29.10 Typical appearance of two individuals with 45,X gonadal dysgenesis. A: This 16-year-old individual has obvious short stature, a webbed neck, shortened fourth metatarsals, and a thoracotomy scar from the repair of the coarctation of the aorta that was performed at 13 years of age. B: This 11-year-old individual also has obvious short stature and stigmata of Turner syndrome. Note that these two individuals look more like each other than they might look like any genetic siblings.
Most 45,X patients have normal intelligence, but many affected individuals have an unusual cognitive defect characterized by an inability to appreciate the shapes and relations of objects with respect to one another (i.e., space-form blindness). Patients with a small ring X chromosome have an increased risk of mental retardation (36). As they grow older, affected children typically are shorter than normal. Although they do not develop breasts at puberty, some pubic or axillary hair may develop because appropriate adrenarche can occur with failure of thelarche (i.e., breast development).
Although less severe short stature and some adolescent development may occur with chromosomal mosaicism, it is reasonable to assume that any short, slowly growing, sexually infantile girl has Turner syndrome until proved otherwise because this disorder is so prevalent (about 1 in 2,500 newborn phenotypic females). In fact, the 45,X karyotype is the single most frequent chromosomal disorder in humans, but most affected fetuses are aborted spontaneously early in pregnancy. However, trisomy is the most common chromosomal type or category of abnormality in first-trimester losses.
The short stature commonly associated with the Turner phenotype appears to result from the loss of a homeobox-containing gene (which encodes for an osteogenic gene) located on the pseudoautosomal region (PAR 1) of the short arms of the X (Xp22) and Y (Yp11.3) chromosomes (37). This gene, which is called either SHOX (short stature homeobox-containing gene) or PHOG (pseudoautosomal homeobox osteogenic gene), escapes X inactivation because of its pseudoautosomal location. The gene appears to account for about two-thirds of the height deficit commonly associated with Turner syndrome.
Even in the presence of typical Turner stigmata, a karyotype is indicated to eliminate the possibility of the presence of any portion of a Y chromosome. Analysis of pooled data suggests that the presence of Y chromosome material is associated with a 12% risk of a gonadoblastoma (38). If a Y chromosome is identified, laparoscopic prophylactic gonadectomy is recommended at the time of diagnosis to eliminate the risk of malignancy. Although gonadoblastomas are benign tumors with no metastatic potential that can arise spontaneously in gonads containing a portion of a Y chromosome, they can be precursors to germ cell malignancies, such as dysgerminomas (most commonly), teratomas, embryonal carcinomas, or endodermal sinus tumors (39). In individuals in whom there is no evidence of neoplastic dissemination, the uterus may be left in situ for donor in vitro fertilization and embryo transfer.
Individuals with Turner syndrome are at increased risk of sudden death from aortic rupture or dissection resulting from cystic medial necrosis during pregnancy, and the risk may be as great as 2% or more (40). In addition, this may occur even if the aortic root diameter is normal. Because of their small stature, ascending aortic diameters of less than 5 cm may represent significant dilatation. Thus, the use of the aortic size index (ascending aortic diameters measured by MRI at the level of the right pulmonary artery normalized to body surface area) is preferred (41). Patients with an aortic size index greater than 2.0 cm/m2 require close cardiovascular surveillance and those with an aortic size index of 2.5 cm/m2 or more are at highest risk for aortic dissection. In fact, the risk of acute aortic dissection is increased by more than 100-fold in young and middle-aged women with Turner syndrome. If pregnancy is being considered, preconception assessment must include cardiologic evaluation with MRI of the aorta.
The evaluation of other commonly involved organ systems should include a careful physical examination, with special attention to the cardiovascular system, and thyroid function tests (including antibody assessment), fasting blood glucose, renal function tests, and intravenous pyelography or a renal ultrasonography.
Treatment of Turner Syndrome
To increase final adult height, commonly accepted treatment strategies include use of exogenous GH (42–44). With recombinant human GH use, the average height gain varied from 4 to 16 cm. It appears that early initiation of therapy (between 2–8 years of age), gradually increasing the dose, and continuing treatment for a mean of 7 years can lead to achievement of a final height greater than 150 cm in most patients (43). Weekly doses of GH of 0.375 mg/kg divided into seven daily doses are typical. Therapy may be continued until a satisfactory height is attained or until little growth potential remains (bone age ≥14 years and growth velocity <2 cm per year). It is not clear if a nonaromatizable anabolic steroid such as oxandrolone will provide additional growth. In girls older than 8 years of age or those with extreme short stature, consideration can be given to using higher doses of GH and adding oxandrolone (45). The dose of oxandrolone should be 0.05 mg/kg per day or less, as higher doses will result in virilization and more rapid skeletal maturation. In addition, liver enzymes should be monitored.
The gonadal steroid treatment of patients with Turner syndrome is as follows:
Mosaic Forms of Gonadal Dysgenesis
Individuals with rare mosaic forms of gonadal dysgenesis may develop normally at puberty. The decision to initiate therapy with exogenous estrogen should be based mainly on circulating FSH levels. Levels in the normal range for the patient’s age imply the presence of functional gonads.
These individuals can become pregnant, with success rates of more than 50% using donor oocytes (48). The increased risk of sudden death during pregnancy resulting from aortic rupture should be assumed to be similar to that of other women with the Turner phenotype (40).
Pure Gonadal Dysgenesis
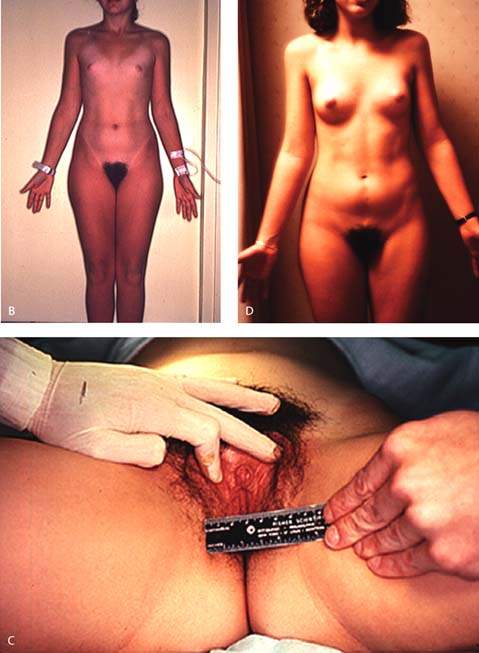
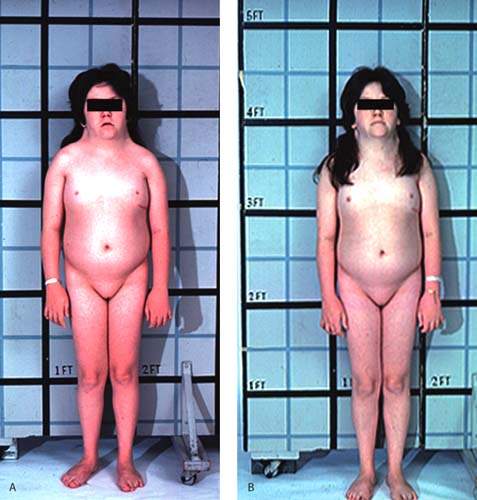
The term pure gonadal dysgenesis refers to 46,XX or 46,XY phenotypic females who have streak gonads. This condition may occur sporadically or may be inherited as an autosomal recessive trait or as an X-linked trait in XY gonadal dysgenesis (Fig. 29.11). Affected girls typically are of average height and have none of the stigmata of Turner syndrome, but they have elevated levels of FSH because the streak gonads produce neither steroid hormones nor inhibin. When gonadal dysgenesis occurs in 46,XY individuals, it is sometimes termed Swyer syndrome. Surgical extirpation is warranted in individuals with a 46,XY karyotype to prevent development of germ cell neoplasms. Both 46,XX and 46,XY forms of gonadal dysgenesis benefit from exogenous estrogen and are potential candidates for donor oocytes.
Figure 29.11 A: A 16-year-old individual with 46,XX gonadal dysgenesis and primary amenorrhea. Circulating follicle-stimulating hormone (FSH) levels were markedly elevated. The small amount of breast development (Tanner stage 2) is unusual, but some pubertal development may occur in such patients. B: A 16-year-old individual with 46,XY gonadal dysgenesis who presented with primary amenorrhea and markedly elevated FSH levels. Most affected individuals do not present with as much pubic and axillary hair development. The right gonad contained a dysgerminoma, but there was no evidence of metastases. (From Rebar RW. Normal and abnormal sexual differentiation and pubertal development. In: Moore TR, Reiter RC, Rebar RW, et al., eds. Gynecology and obstetrics: a longitudinal approach. New York: Churchill Livingstone, 1993:97–133, with permission.) C: Clitoromegaly noted in the girl with 46,XY gonadal dysgenesis depicted in Figure 29.11B. D: The same individual as depicted in Figure 29.11B and D with 46,XY gonadal dysgenesis 1 year after gonadectomy and replacement with exogenous estrogen.