Chapter 57 Principles of Drug Therapy
Pharmacogenetics is the study of how variant forms of human genes contribute to interindividual variability in drug response. The finding that drug responses can be influenced by the patient’s genetic profile has offered great hope for realizing individualized pharmacotherapy when the relationship between genotype and phenotype (disease and/or drug response) predicts drug response (Chapter 56). In the developing child, it is apparent ontogeny has the potential of modulating drug response through altering pharmacokinetics and pharmacodynamics.
General Principles of Pharmacokinetics and Pharmacodynamics
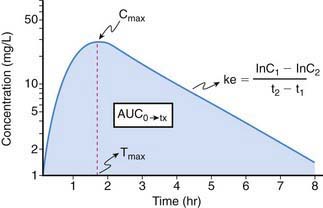
Drug effect is produced only when an exposure (amount and duration) occurs that is sufficient to produce a drug-receptor interaction capable of modulating the cellular milieu and inducing a physiologic response. Thus, exposure-response relationships for a given drug represent an interface between pharmacokinetics and pharmacodynamics that can be simply conceptualized by consideration of two profiles: plasma concentration vs. effect (Fig. 57-1) and plasma concentration vs. time (Fig. 57-2).
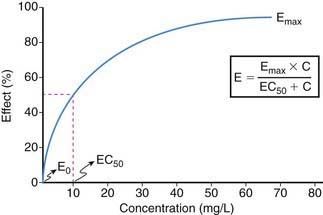
The relationship between drug concentration and effect for most drugs is not linear (see Fig. 57-1). At a drug concentration of 0, the effect from the drug is generally 0 or not perceptible (E0). Following administration of drug and/or escalation of dosage, the concentration increases as does the effect, first in an apparently linear fashion (at low drug concentrations) and then with a nonlinear increase in effect to an asymptotic point in the relationship where a maximal effect (Emax) is attained that does not perceptibly change with further increases in drug concentration. The point in the concentration-effect relationship where the observed effect represents 50% of the Emax is defined as EC50, a common pharmacodynamics term used to compare concentration-effect relationships between patients (or research subjects) and between drugs that may be in a given drug class. In practice, Emax can be derived either from visual interpolation of the concentration-effect profile or via mathematical curve fitting of the relationship.
Because it is rarely possible to measure drug concentrations at or near the receptor, it is necessary to use a surrogate measurement to assess exposure-response relationships. In most instances, this surrogate is represented by the plasma drug concentration vs. time curve. For drugs whose pharmacokinetic properties are best described by first-order (as opposed to zero- or mixed-order) processes, a semilogarithmic plot of plasma drug concentration vs. time data for an agent given by an extravascular route of administration (e.g., intramuscular, subcutaneous, intracisternal, oral, transmucosal, transdermal, rectal) produces a pattern depicted in Figure 57-2. The ascending portion of this curve represents a time during which the liberation of a drug from its formulation, dissolution of the drug in a biologic fluid (e.g., gastric or intestinal fluid, interstitial fluid; a prerequisite for absorption) and absorption of a drug are rate-limiting relative to its elimination. After the time (Tmax) where maximal plasma concentrations (Cmax) are observed, the plasma concentration decreases as metabolism and elimination become rate limiting, the terminal portion of this segment of the plasma concentration vs. time curve being representative of drug elimination from the body. Finally, the area under the plasma concentration vs. time curve (AUC), a concentration- and time-dependent parameter reflecting the degree of systemic exposure from a given drug dosage, can be determined by integrating the plasma concentration data over time.
Definitions of Terms
Area under the curve (AUC) is, conceptually, a measure of both the extent of drug absorbed and its persistence in the body. Thus, it is both concentration- and time-dependent. Mathematically, AUC represents the integral of blood drug levels over time from 0 to either a predetermined postdose time point (AUC0→tx) or extrapolated to infinity (AUC0→∞), which is calculated by using the apparent terminal elimination rate constant (similarly calculated from the observed plasma concentration vs. time plot; see Fig. 57-2).
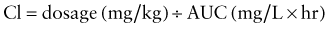
where AUC can either represent the AUC0→∞ for single dose administration or the AUC from time zero to the end of the dosing interval at steady state (i.e., AUCss0→τ). Calculation of Clren requires a complete, quantitative collection of urine (usually over 24 hr) to determine the amount of drug excreted unchanged (Ae). Clnr is generally determined as the difference between Cl and Clren. For drug administration by any extravascular route, the calculation of Cl yields an “apparent” value (e.g., Cl/F) which must be corrected for bioavailability.
Elimination half-life (T1/2) of a drug represents the time for postabsorption blood or plasma concentrations to be reduced by 50%. The apparent elimination rate constant (ke) can be reliably estimated from two drug concentrations obtained on the elimination phase of the concentration vs. time curve (see Fig. 57-2). The T1/2 is then determined as the natural logarithm of 2 divided by the elimination rate constant (T1/2 = 0.693/ke). T1/2 reflects the elimination of parent drug by all of the routes involved in its clearance. Although T1/2 is often considered a surrogate indicator of drug clearance, this should be done cautiously because it depends upon the clearance and the apparent volume of distribution (Vd), as illustrated by the following equation:
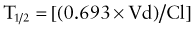
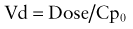
where Cp0 represents the highest attainable plasma concentration following administration of a single dose. As a proportionality constant, Vd is a determinant of plasma drug concentrations attained following administration of a given drug dose. For drugs that do not distribute extensively or associate with great affinity to proteins and tissue, the Vd may dimensionally correspond to a physiologic or anatomic body space: Vd < 0.1 L/kg ≅ intravascular space, 0.1-0.3 L/kg ≅ extracellular space, 0.6-0.7 L/kg ≅ total body water space. Disease and development can influence the Vd, and thus the achievable concentration, for a given drug. Following extravascular drug administration, the apparent Vd is affected by the extent of drug absorbed (Vd/F). In instances in which a drug has incomplete absorption, plasma concentration can underestimate the true value of the Vd.
The Impact of Ontogeny on Drug Disposition
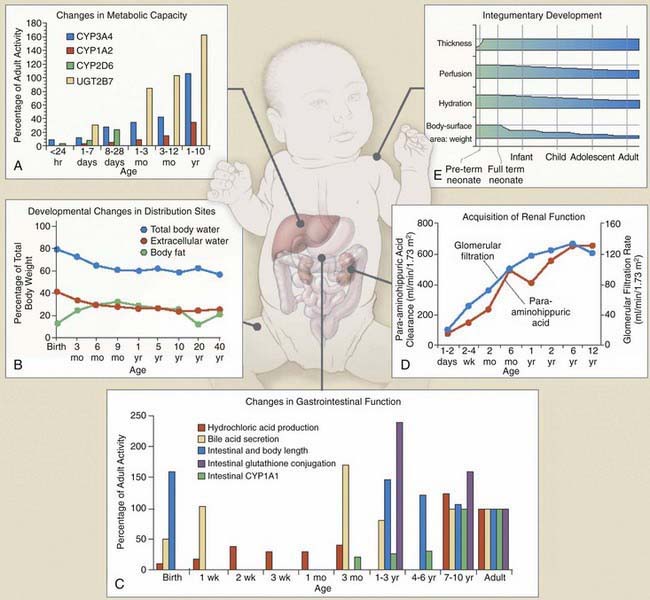
Dramatic pharmacokinetic, pharmacodynamic, and psychosocial changes occur as preterm infants mature toward term, as infants mature through the first few years of life, and as children reach puberty and adolescence (Fig. 57-3). To meet the needs of these different pediatric groups, different formulations are needed for drug delivery that can influence drug absorption and disposition, and different psychosocial issues influence compliance, timing of drug administration, and reactions to drug use. These additional factors must be considered in conjunction with known pharmacokinetic and pharmacodynamic influences of age when developing an optimal patient-specific drug therapy strategy.
Drug Absorption
Peroral Absorption
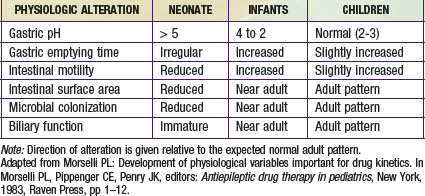
The most important factors that influence drug absorption from the GI tract are related to the physiology of the stomach, intestine, and biliary tract (see Fig. 57-3C and Table 57-1). The rate and extent of peroral absorption of drugs depends primarily on the pH-dependent passive diffusion and motility of the stomach and intestinal tract because both of these factors influence transit time of the drug. Gastric pH changes significantly throughout development, and the highest (alkaline) values occur during the neonatal period. In the fully mature neonate, the gastric pH ranges from 6 to 8 at birth and drops to 2 to 3 within a few hours of birth. However, after the first 24 hr of life, the gastric pH increases due do the immaturity of the parietal cells. As the parietal cells mature, the gastric acid secretory capacity increases (pH decreases) over the first few months of life to reach adult levels by 3-7 yr of age. As a result, the peroral bioavailability of acid-labile drugs, such as penicillin or ampicillin, is increased. In contrast, the absorption of weak organic acids (e.g., phenobarbital and phenytoin) is relatively decreased, a condition that can necessitate administering larger doses in the very young to achieve therapeutic plasma levels.
Extravascular Drug Absorption
Intravenous drug administration is assumed to be the most dependable and accurate route for drug delivery, with a bioavailability of 100%. Absorption of drugs from tissues and organs (e.g., intramuscular, transdermal, and rectal) can also be affected by development (Table 57-2). Intramuscular blood flow changes with age, which can result in variable and unpredictable absorption. Reduced muscular blood flow in the first few days of life, the relative inefficiency of muscular contractions (useful in dispersing an IM drug dose), and an increased fraction of water per unit of muscle mass can delay the rate and/or extent of drugs given intramuscularly to the neonate. Muscular blood flow increases into infancy, and consequently the bioavailability of drugs given by the IM route is comparable to that seen in children and adolescents.
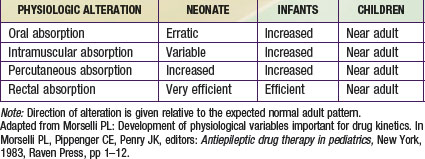
In contrast, mucosal permeability (rectal and buccal) in the neonate is increased and thus can result in enhanced absorption by this route. Transdermal drug absorption in the neonate and very young infant is increased due to the thinner and more hydrated stratum corneum (see Fig. 57-3E). In addition, the ratio of body surface area to body weight is greater in infants and children than in adults. Collectively, these developmental differences can predispose the child to increased exposure and risk for toxicity for drugs or chemicals placed on the skin (e.g., silver sulfadiazine, topical corticosteroids, benzocaine, diphenhydramine), and such toxicity is more likely during the first 8 to 12 mo of life.
Drug Distribution
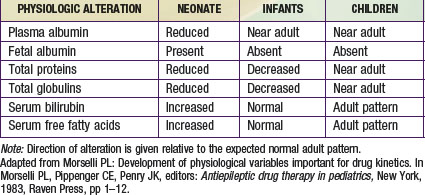
Newborns have a much higher proportion of body mass in the form of water (approximately 75% TBW) than older infants and children (see Fig. 57-3B). Also, the percentage of ECW decreases from the newborn stage (∼45%) into adulthood (∼20-30%). In fact, the increase of TBW in the neonate is attributable to ECW. The reduction in TBW is rapid in the first year of life, with adult values (55%) achieved by approximately 12 years of age. In contrast, the percentage of intracellular water (ICW) as a function of body mass remains stable from the first months of life through adulthood. The impact of developmental changes in body water spaces is exemplified by drugs such as the aminoglycoside antibiotics, compounds that distribute predominantly throughout the extracellular fluid space and have a higher Vd (0.4-0.7 L/kg) in neonates and infants than in adults (0.2-0.3 L/kg).
Albumin, total proteins, and total globulins (e.g., α1-acid glycoprotein) are the most important circulating proteins responsible for drug binding in plasma. The absolute concentration of these proteins is influenced by age, nutrition, and disease (Table 57-3). The concentrations of most all circulating plasma proteins are reduced in the neonate and young infant (about 80% of adult) and reach adult values by 1 year of age. A similar pattern of maturation is observed with α1-acid glycoprotein (an acute phase reactant capable of binding basic drugs) where neonatal plasma concentrations are approximately 3 times lower than in maternal plasma and attain adult values by approximately 1 year of age.
Drug Metabolism
The primary organ responsible for drug metabolism is the liver, although the kidneys, intestines, lungs, adrenals, blood (phosphatases, esterases), and skin can also biotransform certain compounds. Drug metabolism occurs primarily in the endoplasmic reticulum of cells via two general classes of enzymatic processes: phase I, or nonsynthetic, and phase II, or synthetic, reactions. Phase I reactions include oxidation, reduction, hydrolysis, and hydroxylation reactions, and phase II reactions primarily involve conjugation with an endogenous ligand (e.g., glycine, glucuronide, glutathione, or sulfate). As illustrated by Figure 57-3A, many drug-metabolizing enzymes demonstrate an ontogenic profile, with generally low activity present at birth and maturation over a period of months to years (Table 57-4).
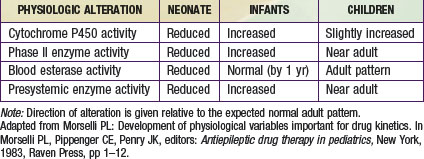
Although there are many enzymes that are capable of catalyzing the biotransformation of drugs and xenobiotics, the quantitatively most important are represented by the cytochrome P450 (CYP450) supergene family, which has at least 16 primary enzymes. The specific CYP450 isoforms responsible for the majority of human drug metabolism are represented by CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A4 (Chapter 56). These enzymes represent the products of genes that in some instances are polymorphically expressed, with allelic variants producing enzymes generally resulting in either no or reduced catalytic activity (a notable exception being the *17 allele of CYP2C19, which conveys increased activity). At birth, the concentration of drug-oxidizing enzymes in fetal liver (corrected for liver weight) appears similar to that in adult liver. However, the activity of these oxidizing enzyme systems is reduced, which results in slow clearance (and prolonged elimination) of many drugs that are substrates for them, including phenytoin, caffeine, diazepam, and many others. After birth, the hepatic CYP450s appear to mature at different rates. Within hours after birth, CYP2E1 activity increases rapidly, and CYP2D6 is detectable soon thereafter. CYP2C (CYP2C9 and CYP2C19) and CYP3A4 are present within the first month of life, a few months before CYP1A2. CYP3A4 activity in young infants can exceed that observed in adults as reflected by the clearance of drugs that are substrates for this enzyme, such as cyclosporine and tacrolimus.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


