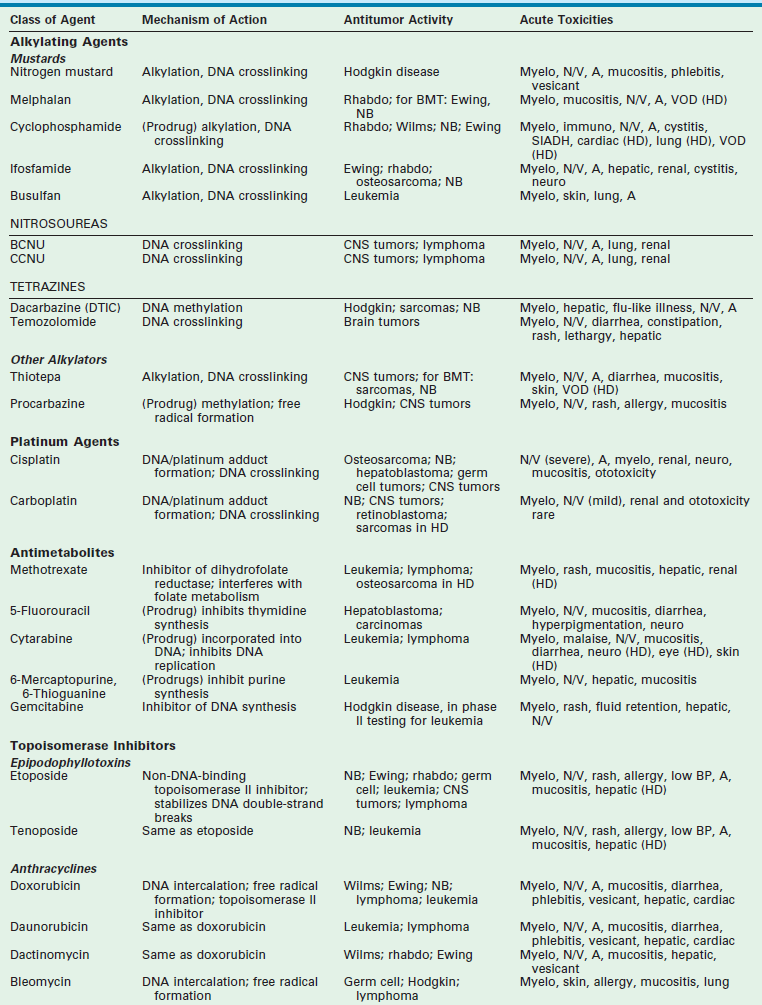
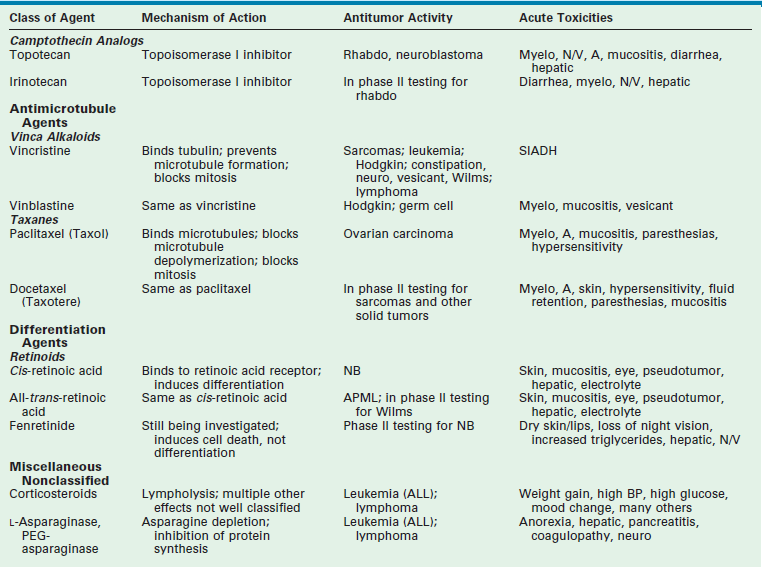
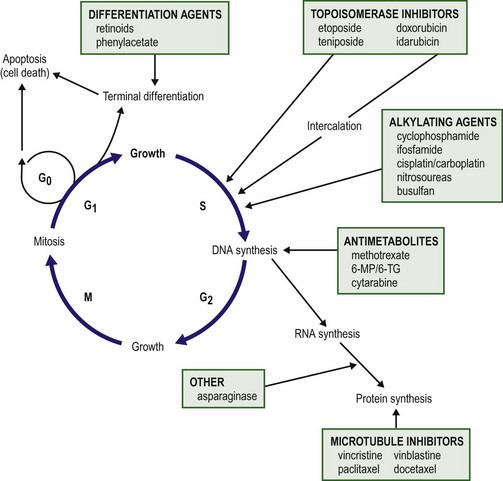
Chapter 64 Early strides in improving oncologic outcomes through the use of single chemotherapeutic agents were first reported by Farber in 19481 and Li in 1956.2 As additional chemotherapeutic agents were developed, they were combined in multidrug regimens that demonstrated both significantly improved response rates and response duration.3,4 By the late 1970s, multimodal therapy was shown to improve cure rates in children with Wilms tumor5 and was being adopted for the treatment of rhabdomyosarcoma, Ewing sarcoma, lymphoma, and other solid tumors.6 The close collaboration of multidisciplinary cooperative groups and the development of improved supportive care measures added impetus to progress. During the 1990s, dose-intensive chemotherapy programs were shown to be successful in improving outcome for patients with Burkitt lymphoma, neuroblastoma, and other advanced-stage solid tumors.7–9 In addition, improvements in outcome were achieved either by alternating effective groups of chemotherapeutic agents to overcome or prevent resistance,10 or by administering agents by continuous infusion rather than bolus.11 By 2001, noncytotoxic biologic therapies (e.g., signal transduction inhibitors, various tissue growth factor receptor inhibitors, antiangiogenesis agents, tumor-targeted antibodies, and adoptive immunotherapy techniques) were developed to target specific biologic pathways.12 In addition, improvements in radiation therapy have led to the development of intraoperative radiation therapy and radiosurgery techniques. Collectively, these advancements have resulted in the continually increasing survival of children with solid tumors and a profound improvement in their quality of life. Although childhood cancers account for only 2% of all reported cancer cases in the general population, they account for 10% of all deaths in children.13 On average, 1 to 2 of every 10,000 children in the USA develop cancer each year.14 The distribution of the types of cancer in childhood is different than in adults. Whereas most cancers in adults have an epithelial cell origin, <10% of childhood cancers fall into this category. The incidence of specific cancers varies by age, gender, and race. Overall, however, it rose from 11.5 cases per 100,000 children in 1975 to 14.8 per 100,000 in 2004.14 The peak incidence (>200 cases per million) is in children younger than 2 years of age. The incidence then decreases to a low of 82.5 cases per million by age 9 years, at which point it begins to rise again through adolescence. In children younger than age 2, central nervous system malignancies, neuroblastoma, acute myeloid leukemia (AML), Wilms tumor, and retinoblastoma account for the majority of diagnoses. In children 2 to 4 years of age, acute lymphoblastic leukemia (ALL) is the most common childhood cancer. After age 9 years, the incidence of Hodgkin disease, osteosarcoma, and Ewing sarcoma begins to increase sharply.13 Over the past several decades, the mortality from childhood cancers has declined dramatically (40%),15 while the 5-year survival rate has risen from 58% in 1975–1977 to close to 80% in 1996–2003.14 Understanding normal cell growth and regulation is a prerequisite to understanding both the genetic basis for the development of childhood cancer and the mechanisms of action of chemotherapeutic agents designed to kill rapidly proliferating cancer cells (Fig. 64-1). Normal cell growth occurs by the regulated progression of the cell through the cell cycle of DNA replication and mitosis. This cycle is separated by two intervening growth phases, referred to as G1 and G2. Cells can temporarily leave the cell cycle and enter a resting state referred to as G0. They are programmed to proceed through the cell cycle by a series of external and internal stimuli. Binding of proteins (growth factors) to cell surface receptors stimulates a cascade of cytoplasmic signaling proteins (membrane kinases and signal transducers) that carries the stimuli to the nucleus. Other proteins (transcription factors) then bind to the DNA, resulting in the expression of growth-regulating genes. When functioning normally, these genes promote or prevent cell division, direct the cell to differentiate, or initiate apoptosis, the process of programmed cell death. FIGURE 64-1 The cell cycle. Normal cell growth proceeds through DNA replication (S) and mitosis (M), separated by two growth phases (G1 and G2). Cells leave the cell cycle to enter a resting phase (G0) to differentiate or to die. Chemotherapy agents act at specific sites along the cell cycle, as indicated. (Adapted from Balis FRM, Holcenberg JS, Poplack DG. General principles of chemotherapy. In Pizzo P, Poplack D, editors. Principles and Practice of Pediatric Oncology. 3rd ed. Philadelphia: Lippincott-Raven; 1997. p. 219.) The presence of consistent cytogenetic abnormalities associated with a specific childhood leukemia or solid tumor is useful in both cancer diagnosis and prognosis. Specific cytogenetic aberrations have been identified in rhabdomyosarcoma, Ewing sarcoma, synovial sarcoma, germ cell tumors, medulloblastoma, neuroblastoma, retinoblastoma, and Wilms tumor.16 Chromosomal aberrations can also be helpful in predicting prognosis. For example, the finding of a chromosome 1q deletion, the presence of double-minute chromatin bodies, or the presence of homogeneous staining regions in neuroblastoma confer a poor prognosis.17 The treatment of Wilms tumor based on risk stratification determined by multiple tumor characteristics that impact prognosis (including loss of heterozygosity at 1p and 16q) is a topic of ongoing study. In the future, specific tumors may be identified by a specific ‘fingerprint’ determined by microarray analysis that can simultaneously analyze expression of thousands of genes on a single chip.18 The ability to tailor therapy to individual patients based on the genetic characteristics of their particular tumor is quickly becoming a reality. Combination chemotherapy remains the mainstay of treatment. The likelihood of cure is maximized when all available active agents are administered simultaneously after local control measures have been undertaken and the tumor burden is as low as possible.19 This approach has led to increased survival rates in children with neuroblastoma, Ewing sarcoma, anaplastic Wilms tumor, and osteosarcoma. The use of adjuvant chemotherapy is supported by the finding that less than 20% of sarcoma and lymphoma patients with initially nonmetastatic solid tumors can be cured by operative or radiation therapy alone or combined.20 Tumor recurrence is generally at a distant site, lending support to the hypothesis that micrometastatic disease exists at the time of presentation for most patients with clinically nonmetastatic disease. As many as 40% of patients with Wilms tumor can be cured with resection or radiation therapy alone. However, survival increases to 90% with the addition of combination chemotherapy.21 As the goal of adjuvant chemotherapy is to prevent the growth of metastatic disease, it is vital that chemotherapy begins as soon as possible after local control measures are employed. For this reason, most current chemotherapy protocols for childhood solid tumors advise that chemotherapy is initiated within 2 weeks of operation.22 In children with Wilms tumor, rapid assessment of tumor biology and risk stratification is important for determining the appropriate chemotherapy regimen. Neoadjuvant chemotherapy has become standard in the treatment of Ewing sarcoma and osteosarcoma, and has the theoretical advantage of minimizing resistance to chemotherapy.10,23,24 Delayed surgical intervention may allow a more complete or less morbid resection, as well as histologic assessment of tumor responsiveness to the chemotherapy agents. Neoadjuvant chemotherapy is beneficial only in tumors for which a known highly effective combination chemotherapy program limits the risk of tumor progression at the primary site. For example, diagnostic biopsy followed by neoadjuvant chemotherapy and delayed resection of the primary tumor for complex neuroblastoma reduces the operative complication rate without compromising survival.25 Advances in supportive care have enabled increased dose intensity of active chemotherapy agents in pediatric clinical trials. These advances decrease or minimize the toxic effects of higher-dose chemotherapy on normal tissues. The use of cytokines (granulocyte colony-stimulating factor [G-CSF] and interleukin-11 [IL-11]) to speed recovery of white blood cells and platelets,26,27 and the use of cardioprotectant agents to allow use of a higher cumulative dose of doxorubicin have helped in the development of new dose-intensive therapy for solid tumors.28 Similarly, progress in bone marrow and stem cell transplantation have allowed dose intensities to be pushed to the upper limits.29–32 The positive impact of increasing the dose intensity on improving response rate and survival has been demonstrated for Burkitt lymphoma, osteosarcoma, Ewing sarcoma, testicular cancer, and advanced ovarian cancer.7–9,33–36 The duration of chemotherapy programs for most pediatric solid tumors has been 1 year. However, as the dose intensity of chemotherapy programs has increased, the duration of therapy has concomitantly decreased. This downward trend is likely to continue. Chemotherapy drugs are divided into classes by their mechanism of action. These classes include alkylating agents (cisplatin and its analogs), antimetabolites, topoisomerase inhibitors, antimicrotubule agents, differentiation agents, miscellaneous nonclassified agents, and biologic agents. Understanding the mechanism of action for each of these agents helps in establishing combination therapies with synergistic antitumor effects. The most common agents from each class, their mechanism of action, common side effects, and tumors in which they are active are listed in Table 64-1. Myelosuppression is an expected side effect of almost all treatment protocols for solid tumors. Transfusions of packed cells and platelets are frequently needed. Of greatest concern is the risk of severe life-threatening bacterial or fungal infections that occur during episodes of neutropenia. In dose-intensive regimens, more than 75% of chemotherapy courses result in hospitalization for fever, with the incidence of bacteremia ranging from 10– 20% per course.37 Success in improving treatment outcomes is partially attributed to advances in supportive care. The routine use of hematopoietic growth factors, specifically G-CSF, results in more rapid granulocyte recovery and shorter hospitalizations for fever and neutropenia.26 In addition, IL-11 enhances platelet recovery, decreases the depth of the platelet nadir, and decreases platelet transfusion requirements.27,38,39 It is well tolerated and is beneficial in combination regimens that induce severe thrombocytopenia.40,41 The gastrointestinal tract is injured by cytarabine, anthracyclines, and high-dose methotrexate; however, the folate derivative Leucovorin can be given to rescue normal mucosal and bone marrow cells from the effects of the latter. No rescue is known for the mucositis and diarrhea which occur with other agents. In addition to enhancing platelet production, IL-11 may help speed recovery from gastrointestinal injury after chemotherapy.42 Renal toxicity can occur from the use of cisplatin, ifosfamide, and high-dose methotrexate. Cisplatin causes renal tubular damage, leading to elevation of levels of blood urea nitrogen and creatinine; this effect is generally reversible. Both ifosfamide and cisplatin cause renal electrolyte wasting in which hypokalemia, hypocalcemia, hypophosphatemia, and hypomagnesemia can occur. Renal injury from these agents can be improved by hyperhydration and forced diuresis. Mesna can prevent hemorrhagic cystitis resulting from cyclophosphamide and ifosfamide by binding to the bladder-toxic acrolein metabolites.40 Additionally, a recent study demonstrates that amifostine administered prior to and during cisplatin infusion significantly reduces the risk of severe ototoxicity in children with average risk medulloblastoma who are undergoing treatment with dose-intense chemotherapy.43 Cellular signaling is a basic biologic function of all normal cells. Signaling can be extracellular (e.g., growth factor receptor tyrosine kinase) or through multiple intracellular effector and survival pathways (e.g., RAS, RAF, TP53, BCL-2). Cancer cells differ from normal cells as they are associated with chromosomal mutations. These mutations result in cancer cells being dependent on several hyperactive signaling pathways. The rationale behind signal transduction therapy is that blocking the hyperactive pathways induces cancer cell apoptosis. By contrast, normal cells have redundant signaling pathways that protect them against cell death. Multiple new signal transduction inhibitors are in development.44,45
Principles of Adjuvant Therapy in Childhood Cancer
Historical Overview
Incidence and Survival Rates
Tumor Biology
Cancer Cytogenetics
Clincial Trials
Adjuvant and Neoadjuvant Chemotherapy
Dose Intensity and Duration of Chemotherapy
Chemotherapeutic Agents
Acute Chemotherapy Toxicity and Supportive Care
Targeting Biologic Pathways
Signal Transduction Inhibitors
Principles of Adjuvant Therapy in Childhood Cancer